Diagnosis of Lafora Disease by Skin Biopsy
Prasath Sathiah1, Debasis Gochhait2, Priyadarshini Dehuri3, Hema Subramanian4
1 Senior Resident, Department of Pathology, Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India.
2 Assistant Professor, Department of Pathology, Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India.
3 Senior Resident, Department of Pathology, Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India.
4 Senior Resident, Department of Pathology, Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Debasis Gochhait, Room No. 2023, 1st Floor, Institute Block, Jawaharlal Institute of Postgraduate Medical Education and Research, Dhanwantri Nagar, Puducherry-605006, India.
E-mail: debasis.go@gmail.com
Periodic acid–Schiff positive inclusions, Seizure, Sweat glands
Lafora disease is an autosomal recessive hereditary brain disorder characterised by recurrent seizures, myoclonus and a decline in intellectual function [1]. This disorder usually appears in late childhood or adolescence and progressively worsens with time [2]. The present case is an 18-year-old female patient who was admitted to the hospital with complaints of seizures since 16 years of age, and it progressively increased for last two years. Subsequently, she developed myoclonus involving leg and progressively decrease in cognitive function. She had a very suggestive family history. Her elder sister suffered from similar complaints at the age of 16 and expired by the time she was 18 years. Her maternal aunt also suffered from mental retardation and expired at 16 years of age.
On clinical evaluation, she did not had any mucocutaneous manifestation. The cardiovascular system was within normal limit. Routine blood examination revealed that patient had iron deficiency anaemia (Hb 8 g/dl). The liver and renal functions were within normal limits, however, serum and urine biochemical tests revealed that patient had hypocalcaemia (6.4 mg/dl) as well as calciuria (38 mg/dl) the cause of which was not ascertained. The Ultrasonography of abdomen and pelvis revealed hepatomegaly but no splenomegaly. MRI of brain did not show any gross anatomical/pathological abnormality. Electroencephalogram (EEG) of brain showed multifocal spikes from right central and frontal region with slow background activity suggestive of generalised epilepsy with encephalopathy. The other serological tests for SLE, HIV, hepatitis B and hepatitis C were negative. The patient had altered sensorium and gradually developed respiratory distress for which she was intubated under general anaesthesia with continuous infusion of antiepileptic drugs (phenytoin). The patient finally succumbed to her illness after eight days in ICU.
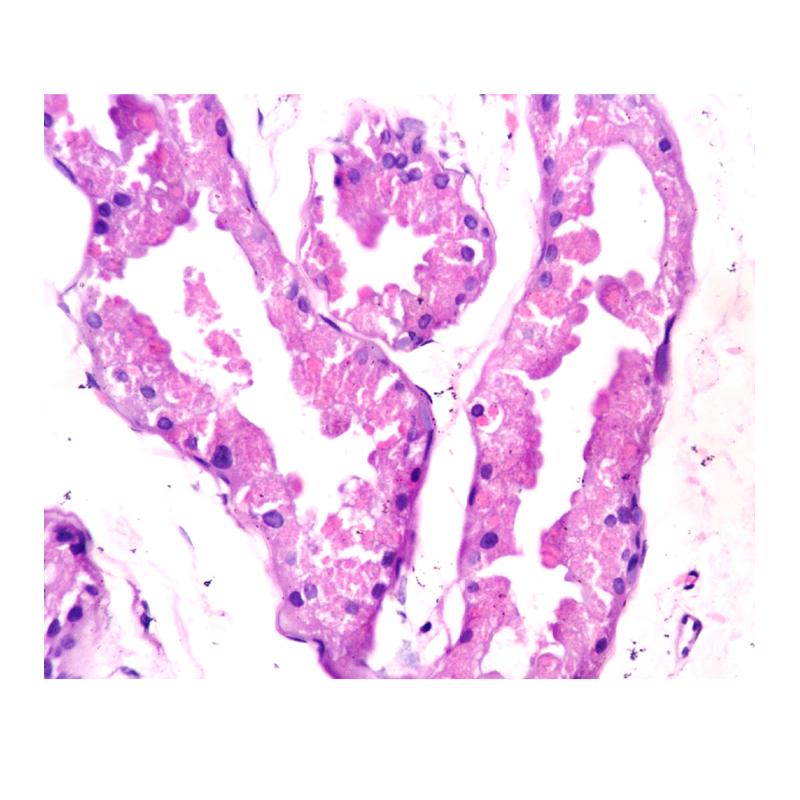
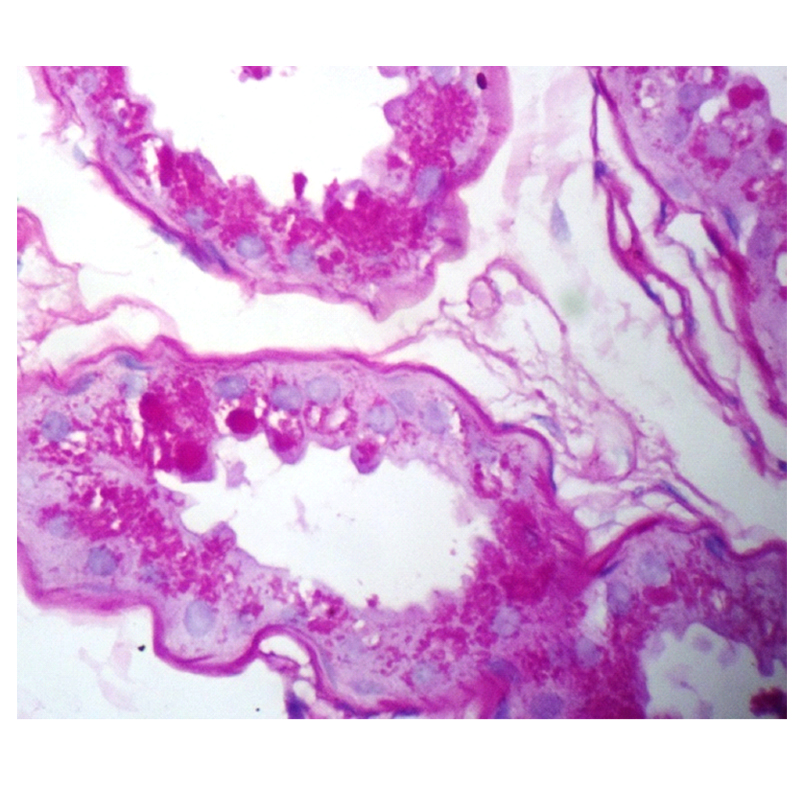
The clinicians suspected Lafora disease as one of the differential for the clinical presentation and a skin biopsy was done from the axillary region. The pathological examination was consistent with clinical suspicion as it showed increase in sweat glands and PAS positive diastase resistant cytoplasmic spherical inclusions in the epithelial cells [Table/Fig-1,2].
Biopsy from axillary region shows intracytoplasmic spherical inclusion in the sweat gland duct (Hematoxylin and Eosin, 40X).
The inclusions are highlighted by PAS stain which were resistant to diastase (PAS, 40X).
Etiology of Lafora disease is unknown [3] and most of these cases present with seizure and develop mental decline in the later course of disease [1]. In the present case also, the patient was presented with seizure and she progressively developed the cognitive decline. The characteristic EEG pattern includes spikes/polyspikes/spike wave complexes in slow background recurrent epileptiform discharges [1]. In the present case also, EEG showed similar pattern in the right central and frontal region. Usually, majority of such patients die between 16 and 24 years of age, six years after the onset of symptoms [1]. The age of death in our case and family history (elder sibling and maternal aunt died around the same age range) was nearly the same as described in literature.
The clinical suspicion of Lafora disease can be confirmed by identification of PAS positive diastase resistant polyglucosan cytoplasmic inclusion bodies in the biopsy of brain (axons and astrocytes), liver, skeletal muscle and axillary skin [4]. The skin biopsy is preferred method because it is less invasive and axilla is the preferred site because it is rich in sweat glands [1]. These inclusions are not specific for Lafora disease as similar inclusions can also be seen in other conditions like in normal aging, Type IV glycogen storage disorder, arylsulfatase A pseudodeficiency, amyotrophic lateral sclerosis and motor neuron disease [5]. One of the normal features of aging is corpora amylacea, which also resembles the Lafora bodies, but it is not found in neuronal structures [6]. The clinical history is very important and helpful for the clinician and pathologist to narrow down on the differentials.
To conclude, many a times the skin biopsy may not have the specific histomorphology, however, a vigilant pathologist should be aware to pick up these inclusions when present in a biopsy to clinch the diagnosis. The prognosis of Lafora disease is very poor and most patients succumb to the disease, however, an early suspicion and axillary skin biopsy may help in an early diagnosis and intervention to prolong the life of these young patients.
[1]. Acharya JN, Satishchandra P, Asha T, Lafora’s disease in South India: A clinical, electrophysiologic, and pathologic studyEpilepsia 1993 34:476-87. [Google Scholar]
[2]. Minassian BA, Lafora’s disease: towards a clinical, pathologic, andmolecular synthesisPediatr Neurol 2001Jul 25(1):21-29. [Google Scholar]
[3]. Carpenter S, Karpeti G, Sweat gland duct cells in Lafora disease: diagnosis by skin biopsyNeurology 1991 31:1564-68. [Google Scholar]
[4]. Nishimura RN, Ishak KG, Reddick R, Porter R, James S, Barranger JA, Lafora disease: diagnosis by liver biopsyAnn Neurol 1980 8:409-15. [Google Scholar]
[5]. Footitt DR, Quinn N, Kocen RS, Oz B, Scaravilli F, Familial lafora body disease of late onset: report of four cases in one family and a rewiev of the literatureJ Neurol 1997 244:40-44. [Google Scholar]
[6]. Rubio G, Garcia Guijo C, Mallada JJ, Cabello A, Garcia Merino A, Diagnosis by axilla skin biopsy in an early case of Lafora’s diseaseJ Neurol Neurosurg Psychiatry 1992 55:1084-85. [Google Scholar]