Primary Intraosseous Paraganglioma (PGL) of sacrum is highly uncommon. Few of the spinal PGL reported were mostly intradural mass. Paraganglionic tissue is usually not present in the bone. So far, only seven cases of primary intraosseous sacral PGL have been reported in the literature. There are no dependable prognostic histological features to differentiate benign from malignant PGL. The only unequivocal criterion for malignancy is metastasis to an organ where paraganglionic tissue is normally not present. However, an aggressive nature can be identified histologically by loss of architecture, decreased or absent sustentacular cells and Ki-67 index of >3%. We report a case of an elderly male who was admitted with complaints of swelling in the lower back with associated radiating pain and difficulty in sitting of two months duration. A diagnosis of sacral chordoma was made on Magnetic Resonance Imaging (MRI). No other mass was detected elsewhere in his body. The patient underwent surgical excision followed by radiotherapy. On histopathology and immunohistochemistry, a diagnosis of locally aggressive primary intraosseous PGL of sacrum was rendered. Hence, when evaluating a lytic sacral mass, PGL has to be considered as a differential diagnosis.
Case Report
A 62-year-old male came with complaints of back pain of two month duration which was insidious in onset, gradually progressive and with difficulty in sitting. Pain was radiating to right lower limb with reduced sensation. He also complained of swelling at the lower back region. No history of bowel or bladder incontinence was given. No history of fever, loss of weight or appetite. He was a known diabetic and hypertensive on tablets metformin 850 mg BD and amlodipine 5 mg OD. On local examination of lower back, a hard and tender swelling of 5 cm x 3 cm in size with ill-defined borders was noticed over the sacral region.
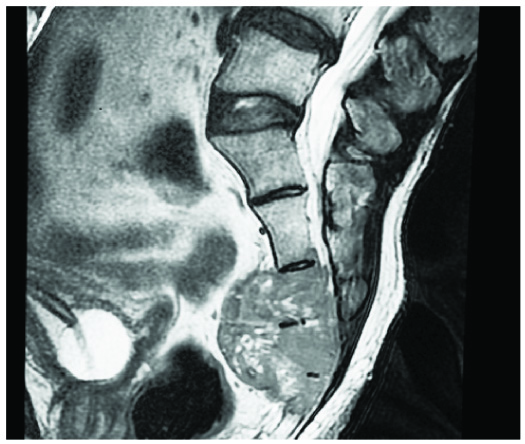
Plain and contrast MRI showed a well-defined lobulated lytic mass lesion which was isointense to muscle on T1, heterogeneously hyperintense on T2 and STIR (short T1 inversion recovery) and showing heterogeneous enhancement involving S2 to S5 sacral vertebrae, their neural foraminae and extending into the presacral space. Imaging features were suggestive of sacral chordoma [Table/Fig-1]. No other mass was detected elsewhere in the body.
MRI image showing a well-defined lobulated sacral mass.
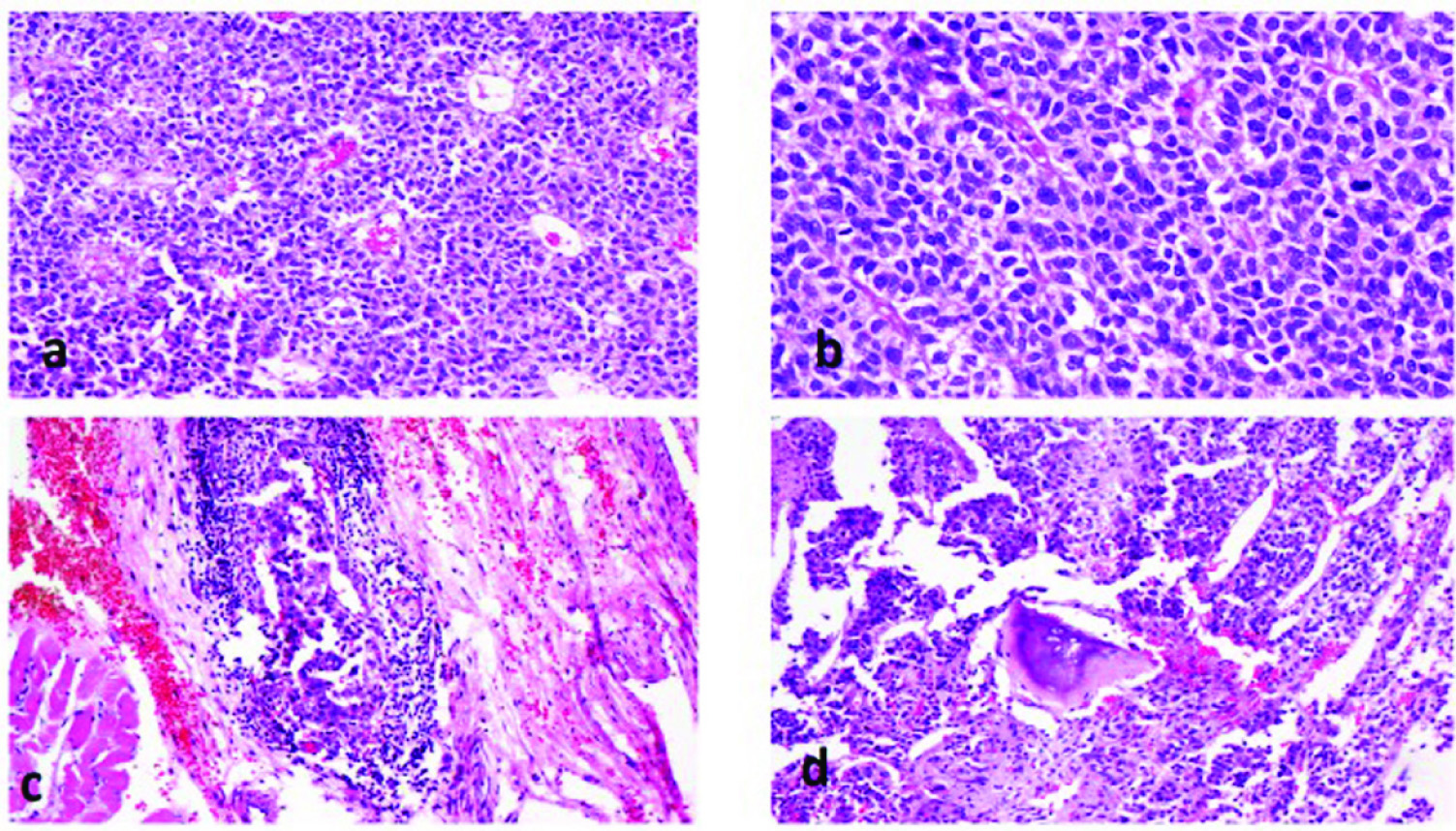
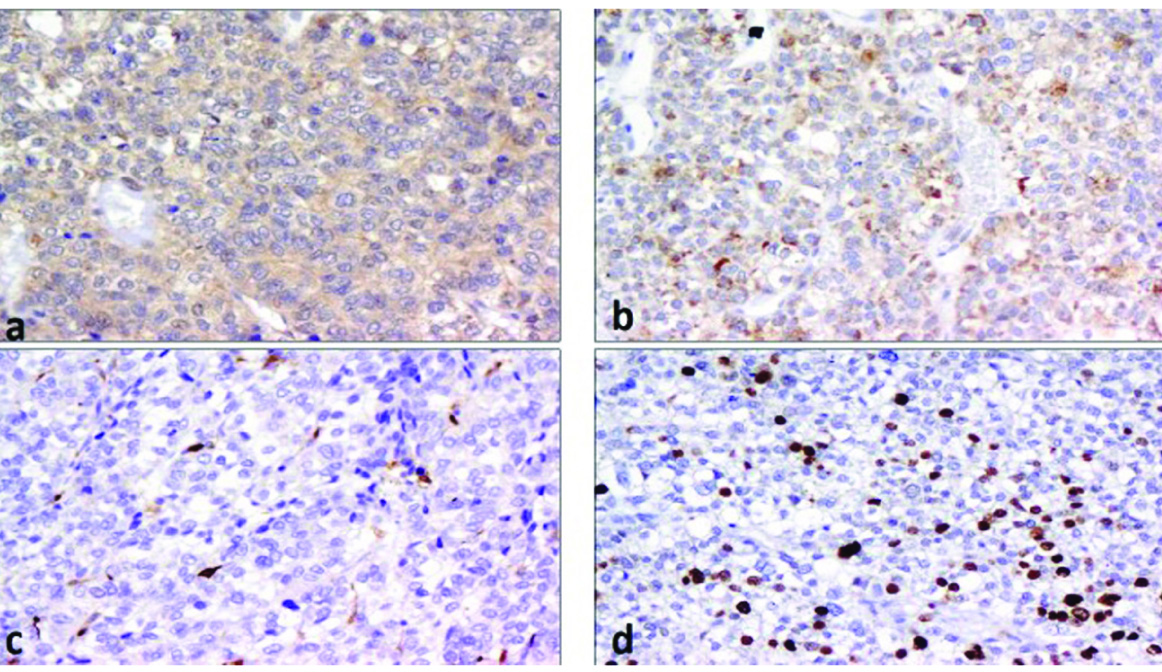
Mass was excised and sent for histopathology. On gross examination, sacrum with an infiltrating, ill-defined, grey white mass measuring 5.5 cm x 2.5 cm x 2 cm was noted. Histological sections showed a tumour comprised of large oval to round cells arranged in an organoid [Table/Fig-2a] and perivascular rosettoid pattern. The tumour cells had moderate amount of pale to eosinophilic cytoplasm, moderately pleomorphic nucleus with irregular nuclear membrane, fine chromatin, small nucleoli, occasional nuclear pseudoinclusions and brisk mitoses [Table/Fig-2b]. Few multinucleated giant cells, small foci of necrosis, vascular emboli [Table/Fig-2c] and bone invasion [Table/Fig-2d] were seen. On immunohistochemistry, the tumour cells showed diffuse mild to moderate expression of chromogranin [Table/Fig-3a] and synaptophysin [Table/Fig-3b] and focally few cells expressed S100 [Table/Fig-3c]. The tumour cells were negative for cytokeratin, Epithelial Membrane Antigen (EMA), vimentin and Glial Fibrillary Acidic Protein (GFAP). Ki-67 index [Table/Fig-3d] was 22%. A diagnosis of locally aggressive primary intraosseous PGL of sacrum was made.
a) Tumour cells arranged in an organoid pattern (H&E,10X); b) Tumour cells showing brisk mitoses (H&E,20X); c) Vascular tumour emboli (H&E,10X); d) Bone invasion by tumour (H&E,10X).
Immunohistochemistry (10X): a) Chromogranin; b) Synaptophysin; c) Focal S100 expression by the sustentacular cells; d) Ki-67.
Postoperative definitive radiotherapy was planned. Patient received 54GY/25 days over six weeks. Patient tolerated well but for grade-2 skin reactions and urinary tract infections. Over the next three months, he developed urinary incontinence and ulcer over the sacral area. In due course, he refused treatment and was discharged at request.
Discussion
Extraadrenal PGL is a rare, highly vascular neuroendocrine neoplasm that arises from autonomic paraganglia. Age of presentation of PGL varies from 13 to 70 years and the mean age of presentation is 47 years [1]. Autonomic paraganglia are small organs which contain neuroendocrine cells derived from neural crest. PGL are derived from either parasympathetic or sympathetic paraganglia [1,2].
Majority of parasympathetic PGL are located in the head and neck region. Carotid body tumours are most common and are not associated with catecholamine secretion or are non-functional. Symptoms result from mass effect [2,3].
Sympathetic PGL arise outside adrenal gland and anywhere along the sympathetic chain from skull base to bladder and prostate. Most of the sympathetic PGL secrete catecholamines, almost always noradrenaline and are called functional tumours. Hence they present with symptoms similar to that of adrenal pheochromocytoma. Some authors have referred to such catecholamine secretion as extraadrenal pheochromocytoma. Functional tumours present with symptoms like episodic headache, palpitation, and sweating. Non-functional tumours present with mass effect. About three-fourth of sympathetic PGL arise in the abdomen, more frequently at the junction of left renal vein and inferior vena cava or at the organ of Zuckerkandl, which is located at the aortic bifurcation near the origin of inferior mesenteric artery [1-3].
Majority of PGL are sporadic and about one-third to one half are associated with inherited syndromes [1]. PGL is an established component of four hereditary syndromes: Multiple Endocrine Neoplasia Types 2A and 2B (MEN 2), Neurofibromatosis Type 1 (NF 1), Von Hippel Lindau (VHL), and the Carney-Stratakis dyad. Germline mutations in genes coding Succinate Dehydrogenase Enzyme complex (SDH D, SDH B and SDH C), VHL, NF-1 and Rearranged during Transfection (RET)[4]. Compared to sporadic PGL, hereditary PGL develops a decade earlier and mostly are multiple at the time of presentation. Sporadic PGL are more common in women, whereas in hereditary PGL, both the genders have equal frequency of occurrence.
Histologically, PGL are composed of round to polygonal epithelioid chief cells arranged in the form of compact nests or trabecular pattern (classical zellballen appearance). Spindly sustentacular or supporting cells are found in the periphery of chief cell nests. The chief cells have moderate to abundant eosinophilic, granular cytoplasm, centrally located nuclei with finely clumped chromatin. In high grade tumours, there is loss of relationship between chief and sustentacular cells with reduced number of sustentacular cells.
There is no reliable prognostic histological marker to distinguish between benign and malignant PGL [3,5,6]. Features of malignancy applicable to other tumours like increased mitotic activity, necrosis, cellular atypia, capsular and vascular invasion have not been found to be reliable [7]. Aggressive behaviour or malignant nature has been linked to loss of architecture of PGL. Sustentacular cells have been reported to be decreased or absent in malignant PGL [8]. A Ki-67 index >3% may be a useful parameter in predicting malignant potential [9]. Ultimately, the presence of metastasis is the sole criteria for malignancy for PGL, more so to a site where paraganglionic tissue is normally absent e.g., liver, bone, lymph node or lungs to avoid confusion with multiple primary tumours [10].
Immunohistochemical markers expressed in chief cells of PGL are Non-Specific Enolase (NSE), synaptophysin, chromogranin, cytokeratin, and vimentin; and in sustentacular cells, S100 and occasionally GFAP are expressed. Some studies have shown that in malignant PGL, there are reduced or absent S100 positive sustentacular cells, and vimentin is expressed selectively in benign PGL and not in the malignant counterpart [10,11].
Rare sites of occurrence of PGL include gall bladder, mesentery, kidney, ovary [12], rectum [11,13], stomach, filum terminale, duodenum, uterus, lung, and cauda equina [14].
Primary intraosseous PGL of sacrum is a rare location of spinal PGL. At the level of sacrum, an intradural extramedullary PGL of cauda equina region has been reported. While extradural spinal PGL were found mainly in the thoracic spine. Coles CP et al., first described intraosseous PGL of sacrum [13]. So far only seven cases of sacral PGL have been reported in the literature [15]. Ours is the eighth case.
The hypothesis for occurrence of PGL in such unusual location like bone is possibly due to more than normal distribution of paraganglia in the foetus. During the embryonic development neural crest cells migrate to unusual sites and differentiate. These can develop into PGL at a later age [15].
Differential diagnosis of a lytic lesion in the sacrum include chondroma, metastatic disease, giant cell tumour, aneurysmal bone cyst, osteoblastoma, chondrosarcoma, lymphoma, haemangio-pericytoma, multiple myeloma, giant sacral schwannoma, myxo-papillary ependymoma and chondroblastoma [14].
Conclusion
PGL are infrequent, highly vascular neuroendocrine tumours arising from extra adrenal autonomic paraganglia. Primary intraosseous PGL of sacrum is a rare occurrence. There is no reliable prognostic histological marker to differentiate benign from malignant PGL. The only undisputable criterion for malignancy is metastasis to an organ where PGL tissue is normally not present. On histopathology, their aggressive nature can be identified by loss of architecture, decreased or absent sustentacular cells and a Ki-67 index of >3%.
[1]. Asad S, Peters-Willke J, Nott L, Malignant paraganglioma, a rare presentation with foot drop: a case reportJournal of Spine Surgery 2015 1(1):99-102. [Google Scholar]
[2]. DeLellis R.A, Lloyd R.V, Heitz P.U, Eng C, World Health Organization Classification of TumoursPathology and Genetics of Tumours of Endocrine Organs 2004 LyonIARC Press [Google Scholar]
[3]. Barnes L, Taylor SR, Carotid body paragangliomas: A clinicopathologic and DNA analysis of 13 tumorsArch Otolaryngol Head Neck Surg 1990 116:44 [Google Scholar]
[4]. Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomasThe Journal of Clinical Endocrinology & Metabolism 2009 94(5):1541 [Google Scholar]
[5]. Kliewer KE, Wen DR, Cancilla PA, Cochran AJ, Paragangliomas: assessment of prognosis by histologic, immunohistochemical, and ultrastructural techniquesHum Pathol 1989 20:29 [Google Scholar]
[6]. Kliewer KE, Cochran AJ, A review of the histology, ultrastructure, immunohistology, and molecular biology of extraadrenal paragangliomasArch Pathol Lab Med 1989 113:1209 [Google Scholar]
[7]. Elder EE, Skog AL, Höög A, Hamberger B, The management of benign and malignant pheochromocytoma and abdominal paragangliomaEur J Surg Oncol 2003 29(3):278-83. [Google Scholar]
[8]. Yang SY, Jin YJ, Park SH, Jahng TA, Kim HJ, Chung CK, Paragangliomas in cauda equina region: clinicopathoradiologic findings in four casesJournal of Korean Neurosurgical Society 2004 35(4):353-58. [Google Scholar]
[9]. Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giache V, Mannelli M, Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paragangliomaJournal of oncology 2012 2012:872713 [Google Scholar]
[10]. Tischler SA, Pheochromocytoma and extra-adrenal paraganglioma: updatesArch Pathol Lab Med 2008 132:1272-84. [Google Scholar]
[11]. Feng N, Zhang WY, Wu XT, Clinicopathological analysis of paraganglioma with literature reviewWorld J Gastroenterol 2009 15(24):300308 [Google Scholar]
[12]. Jahangir N, Shirazi B, Paraganglioma: a diagnostic dilemmaJ Pak Med Assoc 2010 60(9):789-91. [Google Scholar]
[13]. Coles CP, Alexander DI, Gross M, Holness RO, Covert AA, Murray SK, Intraosseous paraganglioma of the sacrum: a case reportCanadian Journal of Surgery 2000 43(2):137 [Google Scholar]
[14]. Wu CS, Wang XW, Qin T, Chen Z, Sun S, Li JM, Primary intraosseous ganglioneuromatous paraganglioma of the sacrum with immunopositivity for cytokeratinEur Rev Med Pharmacol Sci 2015 19(6):931-35. [Google Scholar]
[15]. Kasliwal MK, Rattnani D, Walia BS, Vaishya S, Patir R, Primary intraosseous paraganglioma of the sacrumJournal of Clinical Neuroscience 2011 18(8):1120-22. [Google Scholar]