Congenital limb anomalies are outcome of improper bone formation during embryonic development when cells divide, differentiate with high rate. So, telomerase activity is essential to maintain telomere length for such highly dividing cells. Here, we report four cases of congenital limb anomalies with detailed structures of limbs along with other clinical manifestations of age less than two years. We compared telomere length, expression of telomerase and telomere-associated genes of Peripheral Blood Mononuclear Cells (PBMC) in patient and four age-matched normal individual.
Patient-1 was diagnosed with congenital limb hypogenesis ectrodactyly sequence, an autosomal dominant disorder, showing absence of digits and fibula in upper and lower limb respectively. Both mother and grandmother of Patient-1 showed similar hypogenesis of limbs. Patient-2 showed bilateral clenched hand with arthrogryposis, microcephaly and holoprosencephaly. Both Patient-3 and Patient-4 has no radius in upper limb. Additionally, Paient-3 showed right sided orbital Space Occupying Lesion (SOL) and Paranasal Sinuses (PNS) whereas Patient-4 showed fused kidney with fanconi anaemia. Furthermore, all the patients showed shorter telomere length, inactive telomerase and de-regulated expression of telomere-associated proteins in PBMC compared with age-matched control group.
So, we can conclude that congenital limb anomalies may be linked with telomeropathy and a study with large number of samples is required to firmly establish such association.
CASE-1
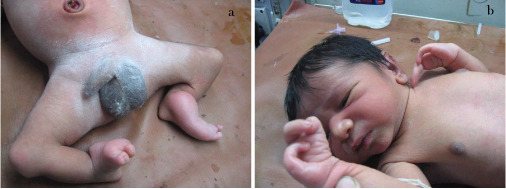
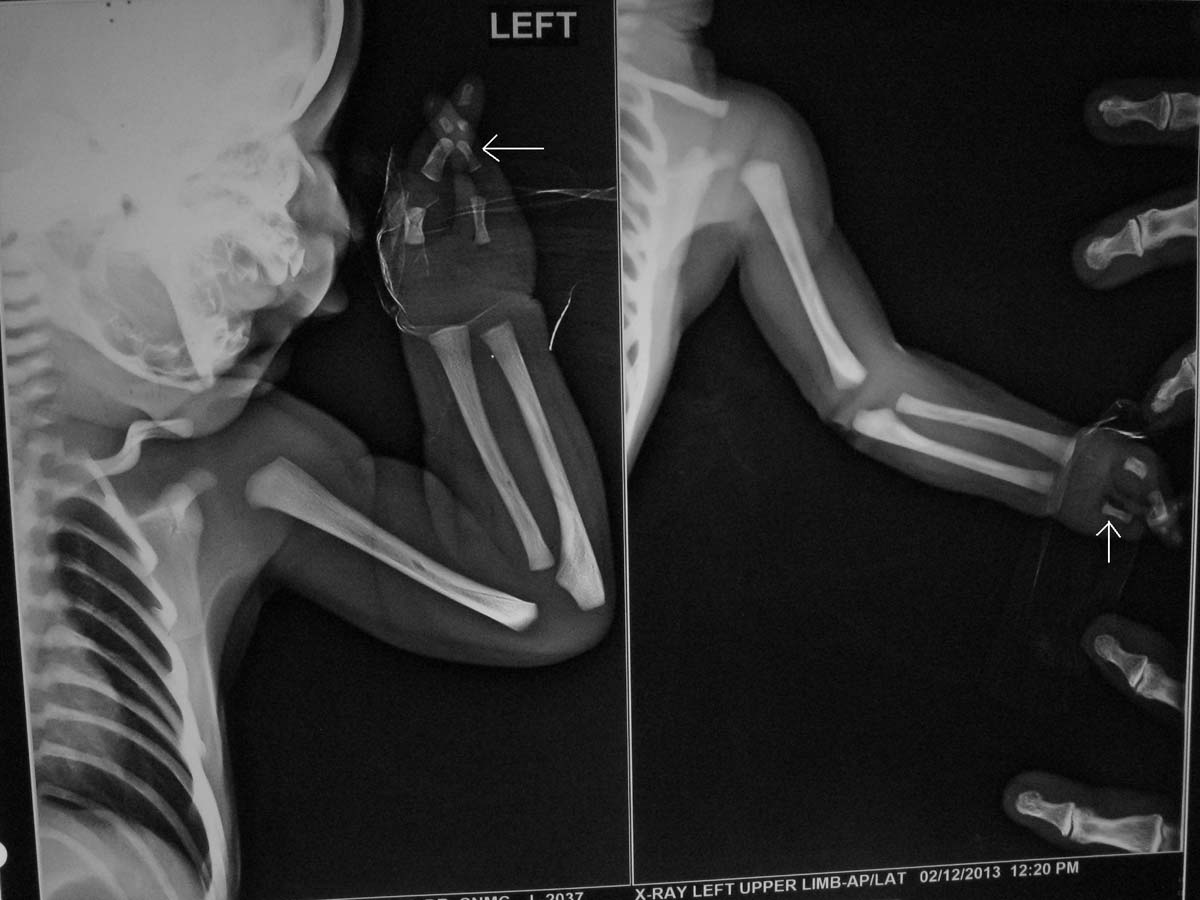
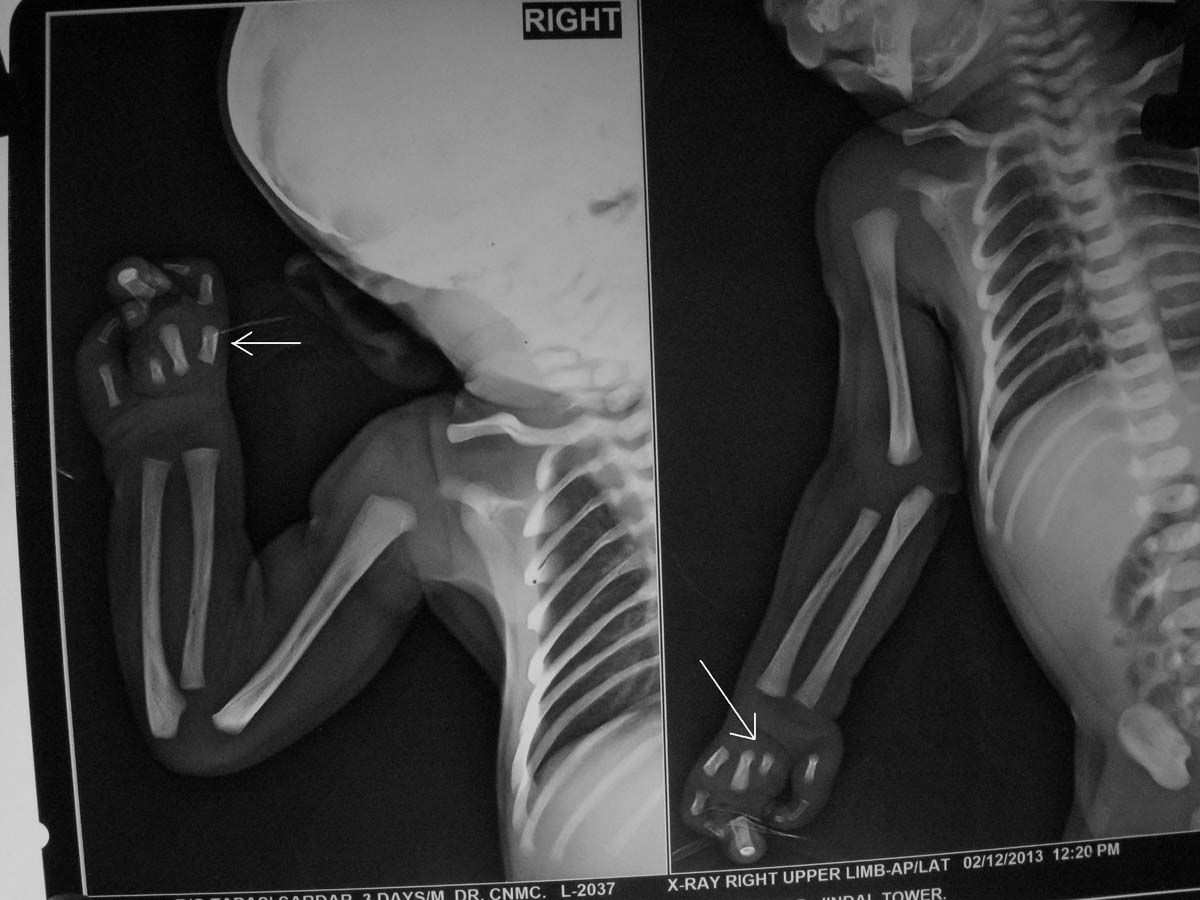
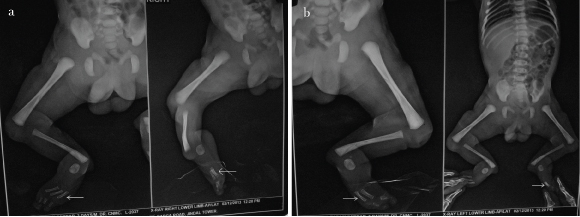
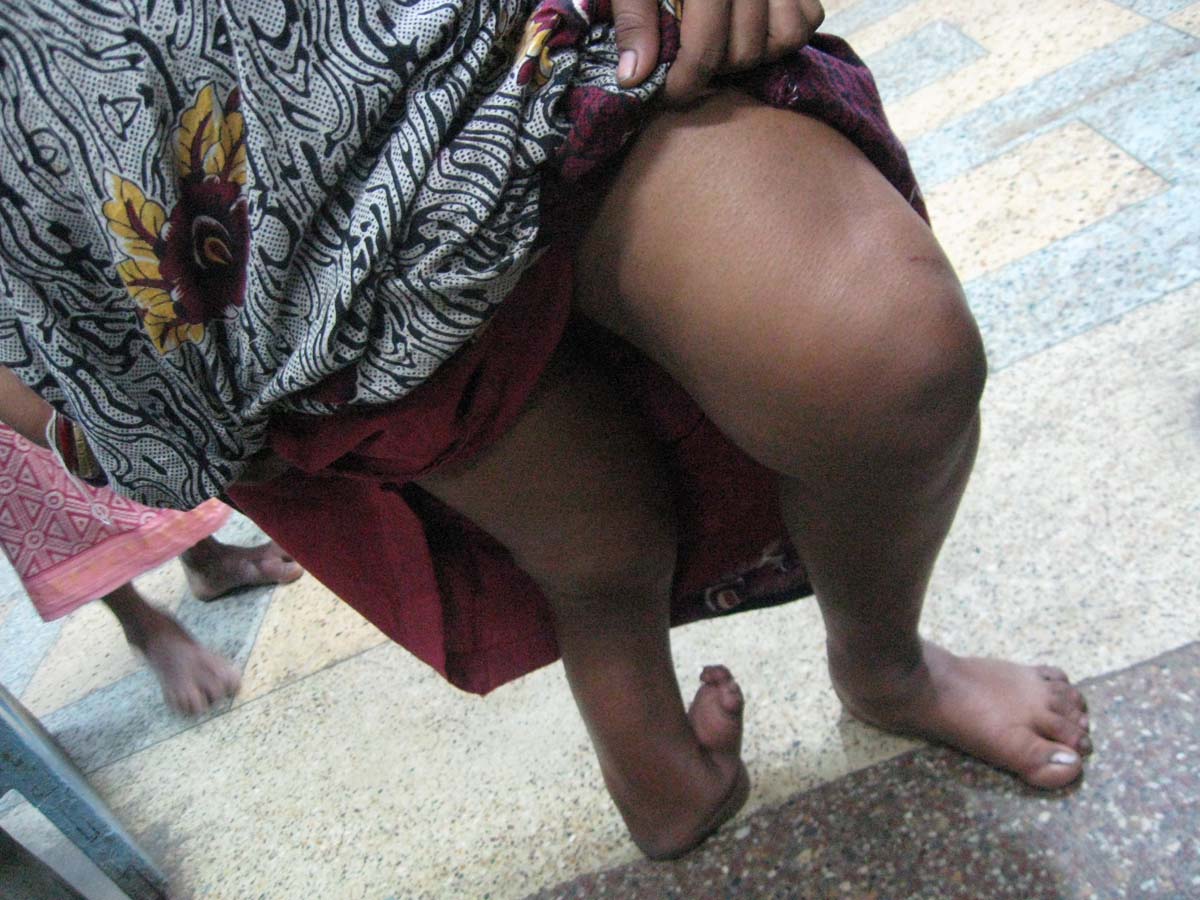
This baby (one-day-old male) presented with bilateral absence of fibula, 3 digits and 2 digits in right and left hands respectively as shown in [Table/Fig-1a,b] {a Split Hand Foot Sequence (SHFS)}. Digital X-Ray showed hypoplasia of limbs with 2 digits and 2 metacarpals in left upper limb [Table/Fig-2] and 3 digits with 4 metacarpals in right upper limb [Table/Fig-3]. X-Ray also showed absence of fibula (only tibia in both legs) with 3 digits in right lower limb [Table/Fig-4a] and 2 digits in left lower limb [Table/Fig-4b]. No other CVS or CNS abnormalities were observed. Interestingly, the grandmother and mother have similar hypogenesis of the limbs as shown in [Table/Fig-5,6] respectively, and also in the maternal cousins. The case was diagnosed to be congenital limb hypogenesis ectrodactyly sequence. It is a limb hypogenesis sequence which occurs in an autosomal dominant fashion in families.
a) Showing fusion of digits with deformity in both lower limbs; b) and showing split hands or ectrodactyly.
X-Ray of left upper limb showing hypoplasia of limbs with 2 digits and 2 metacarpals in left upper limb as labeled by white arrowhead.
XRay of right upper limb showing 3 digits in right hand with 4 metacarpals as labeled by white arrowhead.
X-Ray of lower limbs showing: a) Absence of fibula (only tibia in both legs) with 3 digits in right lower limb as labeled by white arrowhead; b) 2 digits in left lower limb as shown by white arrowhead.
The picture shows hypoplastic fourth and fifth toe in right foot in grandmother of the Patient-1.

The picture shows hypoplastic right lower limb with fusion of the digits in right foot in mother of the Patient-1.
CASE-2
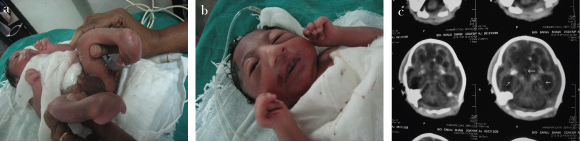
This one-day-old baby (male) also had major degree of limb anomaly with bilateral Congenital Telipes Equinovarus (CTEV) and clenched hands with microcephaly along with genu recurvatum as shown in [Table/Fig-7a,b]. There was presence of holoprosencephaly with cystic white matter degeneration seen in CT scan as shown in [Table/Fig-7c]. This baby was diagnosed to be a case of complex congenital anomaly sequence with arthrogryposis, bilateral clenched hands, microcephaly and holoprosencephaly.
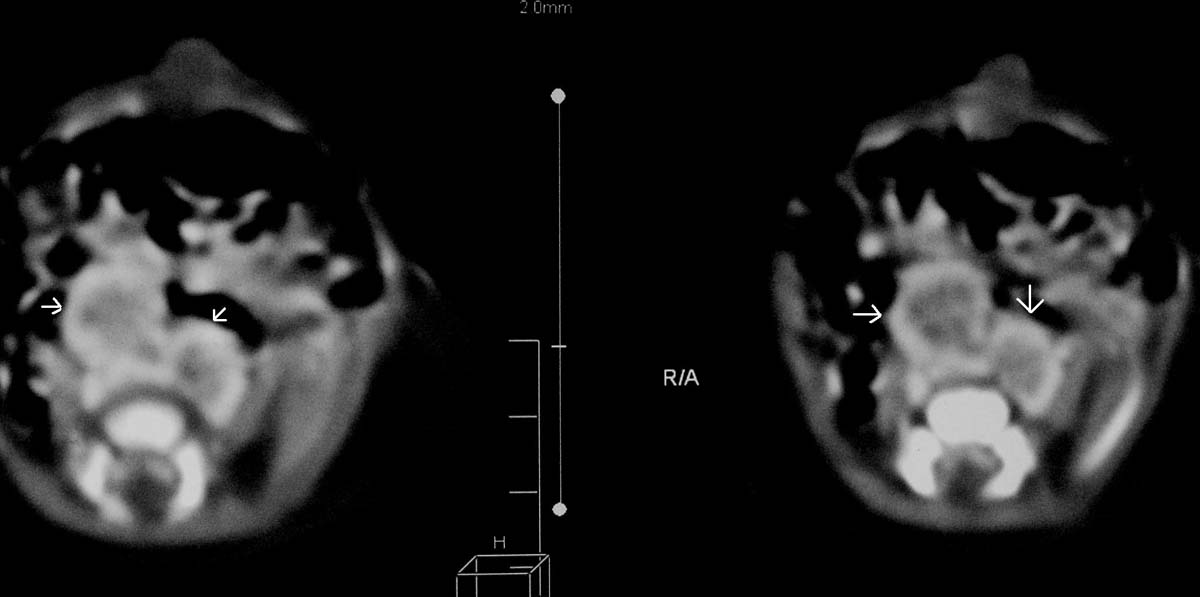
The pictures show: a) Bilateral CTEV; b) Clenched hands; c) Holoprosencephaly as labeled by central thick arrow and cystic changes as labeled by two lateral white arrows in the CT scan of Patient-2.
CASE 3
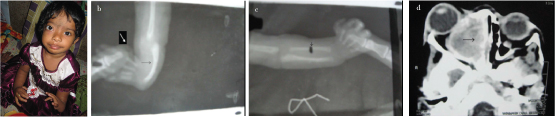
This 18-month-old child (female) had right sided orbital Space Occupying Lesion (SOL) with left sided absent radius as shown by picture of the patient, X-Ray and CT scan orbit and PNS and right sided hypoplastic radius as shown in [Table/Fig-8a-d]. Both the thumbs were attached to hand with a skin tag as shown in [Table/Fig-8a]. As this patient was lost to follow up, we could not perform chromosomal breakage study in this patient and biopsy could not be done from the orbital SOL for histopathological examination. The diagnosis of fanconi was made based on the clinical background. No haematological manifestations were observed.
Normal light image, X-Ray and CT scan of Patient-3: a) Normal photography of the hands of the patient; b) Patient with left sided absent radius a (shown by black arrow head) and absence of first metacarpal (thumb attached with skin tag) shown by white arrow heads in X-ray image; c) Right radius is present but hypoplastic (black arrowhead) in X-ray image; d) CT scan shows orbit and PNS (paranasal sinuses) showing SOL in the right maxillary antrum as labeled by black arrow.
CASE-4
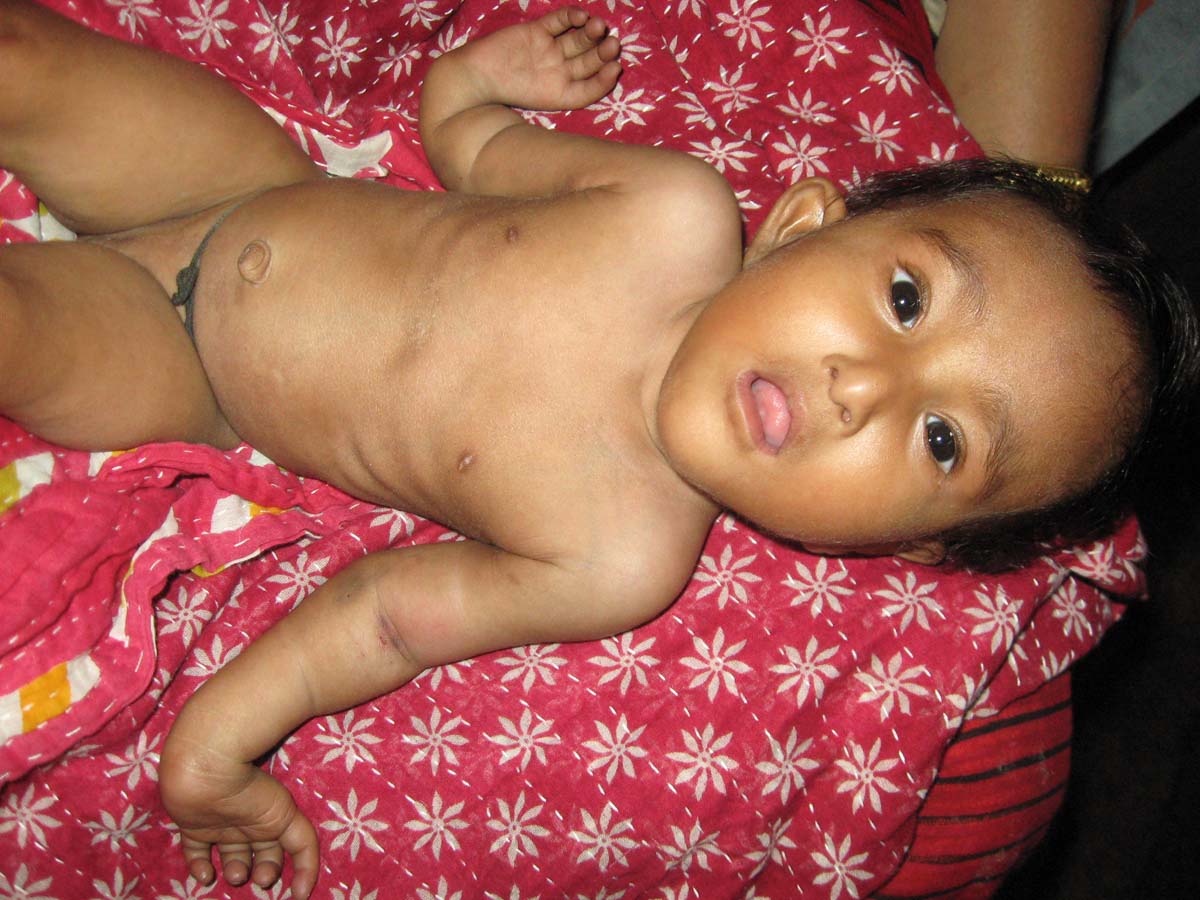
This was an eight-month-old female child, presented with hyper-telorism, short stature and gradually evolving hyperpigmentation of whole skin as shown in [Table/Fig-9]. She had right sided absent radius and 4 digits in each hand with bilateral absent thumb as shown in [Table/Fig-10]. Additionally, she had fused kidneys in abdominal as detected by CT scan as shown in [Table/Fig-11]. Significant chromosomal breakage was observed in mitomycin-c treated PBMC of this patient compared with untreated PBMC (data not shown), implicating that the patient was Fanconi positive. Furthermore, the patient had trisomy 3 (data not shown).
The Patient-4 shows bilateral absent thumb with right sided radial ray defect.
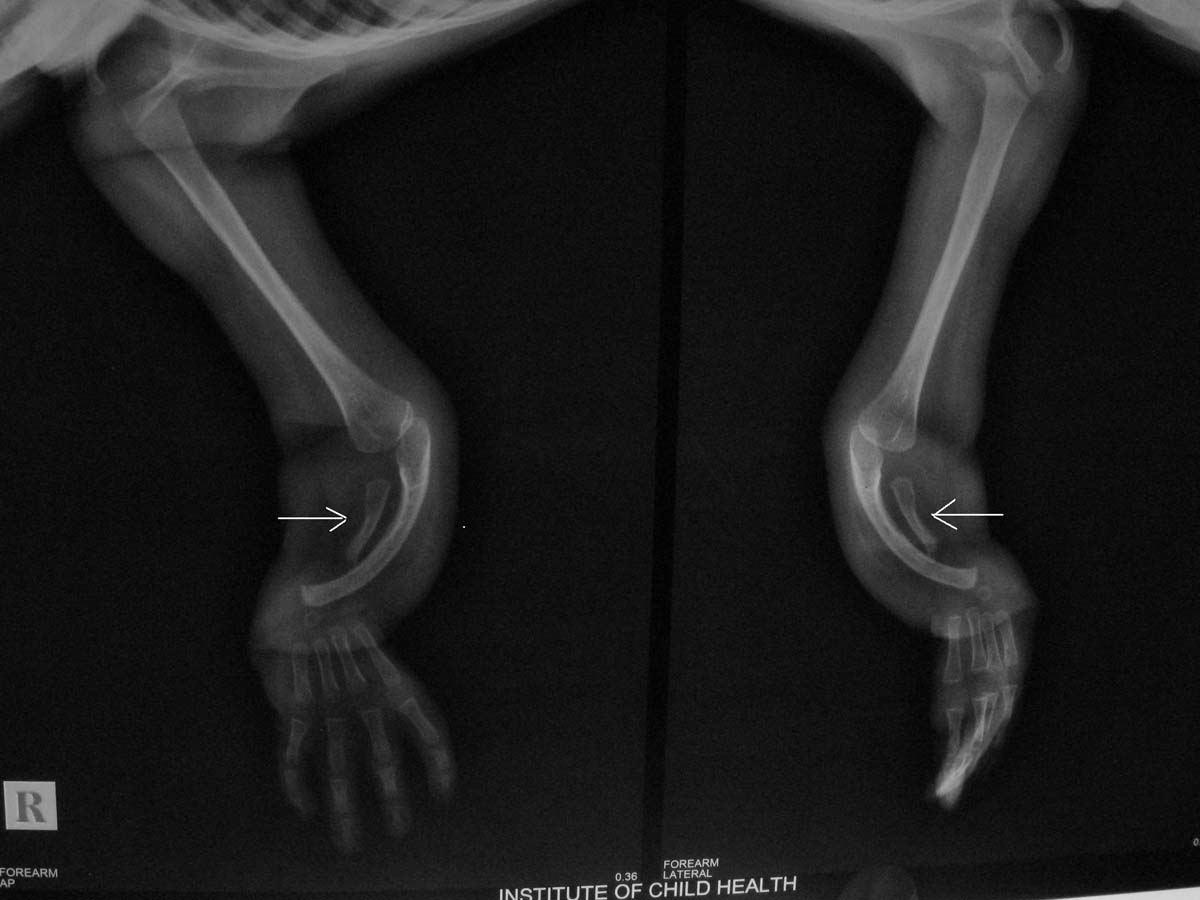
X-Ray picture of hands of Patient-4 showing bilateral small hypoplastic radius as labeled by white arrowhead and absent thumb (4 digits in hand) with proved Fanconi.
CT abdomen of Patient-4 shows fused kidneys as labeled by white arrowhead.
Discussion
Very little is known about regulation of embryonic development and it is a very complex network of signaling pathways where a large number of genes are involved. So far a set of genes are identified those are associated with proper limb growth or development [1,2]. During the limb development, proper bone formation must be associated with a working vascular network to supply nutrients to the cells through blood. So, it is expected that the genes involved in angiogenesis such as VEGFA, HIF1A, NOS3 are involved in bone development [3]. Telomerase activity is urgently needed to maintain telomere length during embryonic development due to high rate of cell division [4]. In fact, there is regulation of tissue-specific expression and activity of telomerase in differentiated cells during various stages of gestation in human as well as in mouse [5,6]. Thus, proper embryonic development is linked with telomerase and telomere biology. Telomere length erosion can be reversed by telomerase holoenzyme or by telomerase-independent ALT pathway where a large number of telomere-associated proteins are involved and any deregulation of these proteins result telomere dysfunction, impaired ALT pathway and short telomere [7–9].
At first, four patients [Table/Fig-12] with limb anomaly are chosen according to the common criteria of having limb anomalies (both upper and lower) from a group of patients of age group 0-2 years, presenting at paediatric outpatient Department of Calcutta National Medical College and Hospital (CNMC & H). Blood samples were collected during out patients care unit of CNMC & H, Kolkata, India, strictly following the prescribed protocol of Institutional Ethical Committee. The patients were examined clinically and after suspecting the clinical diagnosis we processed the blood for telomere length measurement and expression of telomere proteins. We have age-matched four normal individual (clinically normal infants of same age with normal phenotype) and termed as control group. Then RNA was isolated from whole blood and cDNA was prepared for gene expression studies by real time PCR.
The significant features of the four cases.
| Patient serial no: | Clinical features | Radiological features | Diagnosis | Telomere length and telomerase activity | Expression of telomere associated proteins |
|---|
| CASE 1 | Split hand and leg digits (Autosomal dominant inheritance in family) | X-Ray shows 2 digits left upper limb [Table/Fig-2] and 3 digits in right upper limb [Table/Fig-3]. Absence of fibula with 3 digits in right lower limb and 2 digits in left lower limb lower limb as shown in [Table/Fig-4a,b] | Congenital limb hypogenesis ectrodactyly sequence | Shortened telomere and dysfunctional telomerase with complete shutdown of hTERT and hTERC gene. | Significant reduction of RAP1, TPP1, POT1, TERF1 and abnormal high expression of TERF2 and BTBD12 |
| CASE 2 | Bilateral CTEV, with clenched hands and microcephaly | CT brain shows holoprosencephaly [Table/Fig-7c] | Holoprosencephaly with clenched hands linked to SHH gene on chr 7q | Shortened telomere and dysfunctional telomerase with complete shutdown of hTERT and hTERC gene. | Almost all proteins under study are significantly reduced. |
| CASE 3 | Bilateral radial ray defect with right orbital SOL | X-Ray shows right sided hypopalstic radius and left sided absent radius with absent first metacarpals [Table/Fig-8c,d]. CT orbit and PNS shows right sided SOL. | Probable Fanconi as stress test not performed and diagnosis was on clinical background | Shortened telomere and dysfunctional telomerase with almost complete shutdown of hTERT and hTERC gene. | TERF1 and RAP1 severely reduced while POT1 and PARP-1 unusually enhanced. |
| CASE 4 | Hypertelorism, fused kidney with bilateral radial ray defect and absent thumb bilaterally. | X-Ray showed right sided absent radius with bilateral absent thumb (4 digits in each hand) along with fused kidney in CT abdomen [Table/Fig-10,11]. | Confirmed Fanconi (stress test positive showing increased chromosomal breakage) | Shortened telomere and dysfunctional telomerase with almost complete shutdown of hTERT and hTERC gene. | TERF1, TPP1 and RAP1 severely reduced while TERF2 and POT1 usually increased. |
PBMC were extracted from the whole blood cells using ficoll (Invitrogen) following standard protocol. The extracted PBMC were kept at -80°C in PBS for further downstream experiments.
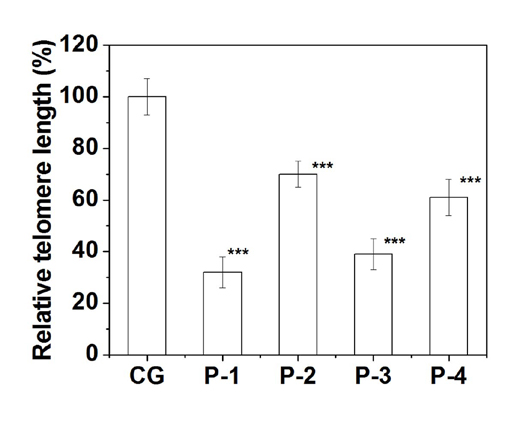
Relative telomere length was measured by qPCR technique using genomic DNA from the isolated PBMC [10]. The relative T/S ratio was calculated using albumin as an endogenous control. The relative telomere length in PBMC of all the patients in comparison to age-matched normal individual group is shown in [Table/Fig-13]. All the patients showed a significant (p≤0.001) shorter telomere length than control group and it ranged from 40% to 70% reduction of telomere length. This data implicates that all four congenital limb anomalies patients in our study show compromised telomere length maintenance pathway.
Relative telomere length (%) of all the patients are shown here. Telomere length of each patient was measured by real PCR and compared with mean telomere length of control group. Here ‘***’ denotes p≤0.001.
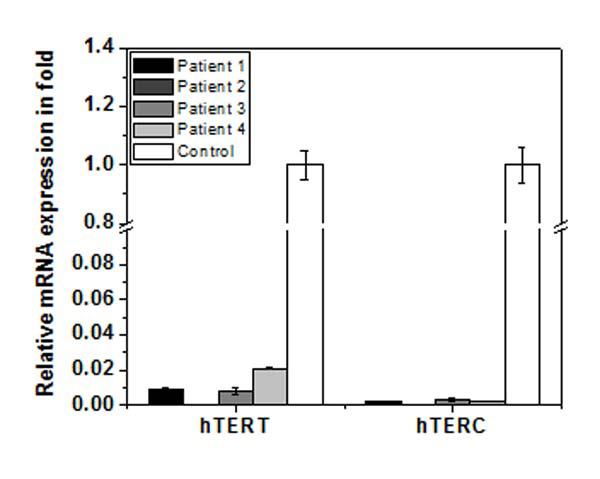
To search for the reason behind the shorter telomere length of the patients, we monitored expression of two key subunit of telomerase - hTERT and hTERC in whole blood cells from patients and age-matched control group using real time PCR. The data is shown in [Table/Fig-14]. The expression of hTERT in all the patients was below 4% compared with age-matched control group and in some patients it was practically zero. Similarly, the expression of hTERC was below 1% of control group. This data implicates that hTERT and hTERC are significantly reduced in patients and hence telomerase activity is expected to be significantly low or zero. Thus, shorter telomere in patients may be due to zero or very low telomerase activity.
Expression of hTERT and hTERT gene of whole blood of each patient as measured by real time PCR. The expression of each gene of each patient is compared with mean expression of that gene in age-matched control group (hollow bar). Expression of both the genes in each patient is significantly (p≤0.001) reduced.
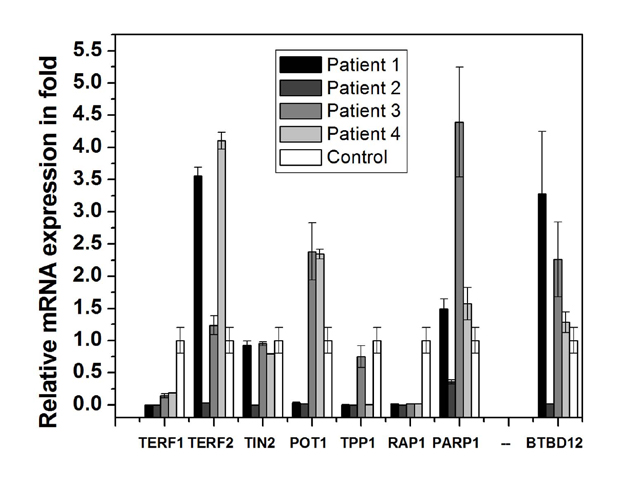
We have checked expression of six shelterin complex and two non-shelterin proteins BTBD12 and PARP-1. The expression all eight telomere-associated proteins are deregulated differentially in different patients as shown in [Table/Fig-15]. Patient-1 showed significant reduction of RAP1, TPP1, POT1 and TERF1 whereas significant increase of TERF2 and BTBD12 compared with control group. Patient-2 showed almost all the proteins are significantly reduced to almost zero. Patient-3 and 4 showed severely reduced TERF1, TPP1 and RAP1 while the other proteins are almost same or slightly higher. Such type of deregulation of telomere-associated proteins results telomere dysfunction which may result short telomere and chromosomal aberration.
Expression pattern of shelterin and non-shelterin (PARP-1, BTBD12) telomere-associated proteins of whole blood of each patient as measured by real time PCR. The expression of each gene of each patient is compared with mean expression of that gene in age-matched control group (hollow bar).
The patient-1 showed congenital limb hypogenesis ectrodactyly sequence or SHFM1. It is a limb developmental disorder characterized by missing digits, fusion of remaining digits, and a deep median cleft in the hands and feet (also known as erectodactyly), and is a genetically heterogeneous human disorder with autosomal dominant inheritance pattern [11]. The SHFM1 critical region has been mapped on the basis of chromosomal rearrangements to ~1.5-Mb interval on 7q21.3 [12]. Till now no disease gene has been identified. However, three candidate genes such as DSS1, the homeobox genes DLX5 and DLX6 are located in this region.
The Patient-2 showed the congenital anomaly with holoprosen-cephaly and limb anomaly (arthrogryposis and clenched hand) which is linked to SHH gene on chromosome 7q. Most cases of holoprosencephaly type 3 (HPE3) is caused by deletions and point mutations in the Sonic hedgehog (SHH) gene on chromosome 7q36 itself, but can also be caused by translocations up to 265 kb upstream of the gene [13]. Phenotypic expressivity is variable, ranging from a single cerebral ventricle and cyclopia to clinically unaffected carriers in some familial HPE cases. Overall the limb anomalies in human development are strongly controlled by HOX gene cluster in addition to the individual genes discussed above. Interestingly, there is a position effect of these genes on the anatomical development of the limb bud. An elegant series of deletion and duplication experiments in mouse [14] has demonstrated that an early limb bud activating element (ELCR) lies telomeric to the HoxD cluster, an idea first hypothesized by Zákány J et al., [15]. The hypothesis says: if a gene is positioned closer to the telomeric end of the cluster, either naturally or by artificial means, it is activated earlier during development and it is expressed within the limb bud more anteriorly. Due to financial constraints we could not study the genetic confirmatory tests for Patient 1 and Patient 2.
Fanconi pancytopenia syndrome has an autosomal recessive inheritance pattern and this disease can be diagnosed by chromosomal breakage analysis induced by MMC (stress test) in peripheral blood leucocytes [11]. Patient-4 in our study also had Fanconi anaemia. Clinical abnormalities include short stature, microcephaly in 25%-37% cases, mental retardation in 25% cases, eye anomalies, radial ray defect in 49% cases including hypoplasia or aplasia of thumb, hypoplastic or absent radii, renal and urinary tract anomalies in 34% cases including malformed kidneys and double ureters [11]. Haematologic manifestations develop usually in the first decade with progressive bone marrow failure and development of malignancy, especially AML and to lesser extent solid tumours. Patient-3 in our study has developed solid tumour of the right orbit and we diagnosed the patient clinically to be a probable case of fanconi having radial ray defects bilaterally. Interestingly, there exists a marked variability of the clinical phenotype [16]. About 25% of affected individuals are structurally normal but are diagnosed as fanconi cases due to presence of pancytopenia and positive DNA breakage analysis. Since the median age of the haematologic abnormalities is around first decade, this characteristic dysmorphic features (even in the absence of haematologic malignancies) as diagnosed at early stage could give better opportunity to the patient to survive by Haemopoietic Stem Cell Transplant (HSCT) from a HLA matched sibling malignancies [17].
We did not study the critical region of chromosome 7 or Hox gene cluster which are reported to be associated with limb anomalies as discussed above. However, we have monitored telomere length and expression of few genes involved in maintaining telomere length. Here, we see that all the patients show shorter telomere length compared with age-matched control group. The reason behind such shorter telomere is inactive telomerase and dysfunctional telomere due to the deregulated telomere-associated proteins. A number of diseases or disorders are consequences of the defects in telomere maintenance machinery such as dyskeratosis congenital [18], Idiopathic Pulmonary Fibrosis (IPF) [19], anaemia with or without bone marrow failure [20,21], liver cirrhosis [22].
The impaired telomere maintenance machinery is also termed as telomeropathies and it is found in many more disorders [23]. But, there are no reports of association of this telomeropathies in congenital limb anomalies. However, association of telomerase/telomere length maintenance with proper embryonic development is an established fact in human as well as in mouse [5,6,24]. Both the hTERT and hTERC are required for osteoblastic bone formation and osteoblast differentiation implicating that telomerase activity and telomere length maintenance is urgently needed in proper bone formation in embryo [25,26]. Furthermore, telomerase activation by over-expression of hTERT activates IGF-AKT signaling which gets impaired upon deficiency of telomerase leading to reduced bone formation [25]. Thus, absence or malformation of bones in both upper and lower limbs as seen in our patients may be due to inactive telomerase.
Role of PARP-1 is not clear. However, it can alter expression of several genes via modulating transcription factors such as NF-κB, B-MYB, Oct-1 during various stages of embryogenesis [27]. PARP-1 is reported to have role in implantation of embryo through preparation of uterus [28–31]. In our study, PARP-1 was significantly reduced in patient-2 only and increased in the rest of the patients. Such de-regulated PARP-1 may have role in improper limb development. BTBD12 is a well-known structure-specific endonuclease and important TRF2-interacting partner, has role in recombination repair and telomere stabilization [32,33]. BTBD12 is reported to have role in mammalian gametogenesis as it can control the repair process [34]. This protein is significantly reduced in Patient-2 and increased in other patients. So, we cannot rule out its function in proper limb development.
Conclusion
To the best of our knowledge this is the first report of patients with congenital limb anomalies also suffering from telomeropathy. Here, we caution that we have studied only four patients and a detailed investigation is required in a large number of patients in future to confirm the association of telomeropathy with congenital limb anomalies, if any.
Financial support: This work was partially supported by funds received by UG from DST (SR/SO/BB-0017/2010), New Delhi, Government of India and DBT (BT/PR 4809/BRB/10/1028/2012), New Delhi, Government of India.
AUTHOR CONTRIBUTION
JM collected samples and did various clinical test like X-rays, CT scan etc. TB assisted JM in diagnosis and such clinical tests. Both JM and TB did all these clinical jobs under supervision of BCM.
Isolation of DNA and RNA from blood was done by PC and all the gene expression profile, telomere length was measured by PC.
Considering the work load, the authors JM and PC contributed equally to this work.
UG designed the whole project proposal and all molecular experiments. Funds received by UG was partially utilized to carry out all molecular experiments done by PC. The manuscript was drafted by UG taking the help of JM in clinical aspects.
[1]. Barham G, Clarke NMP, Genetic regulation of embryological limb development with relation to congenital limb deformity in humansJournal of Children’s Orthopaedics. Epub ahead of print 2008 [Google Scholar]
[2]. Johnson RL, Tabin CJ, Molecular models for vertebrate limb developmentCell 1997 90:979-90. [Google Scholar]
[3]. Gregg AR, Schauer A, Shi O, Liu Z, Lee CG, O’Brien WE, Limb reduction defects in endothelial nitric oxide synthase-deficient miceAm J Physiol 1998 275:H2319-24. [Google Scholar]
[4]. Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW, Telomerase activity in human germline and embryonic tissues and cellsDev Genet 1996 18:173-79. [Google Scholar]
[5]. Ulaner GA, Giudice LC, Developmental regulation of telomerase activity in human fetal tissues during gestationMol Hum Reprod 1997 3:769-73. [Google Scholar]
[6]. Prowse KR, Greider CW, Developmental and tissue-specific regulation of mouse telomerase and telomere lengthProc Natl Acad Sci USA 1995 92:4818-22. [Google Scholar]
[7]. Blackburn EH, Switching and signaling at the telomereCell 2001 106:661-73. [Google Scholar]
[8]. Kamranvar SA, Chen X, Masucci MG, Telomere dysfunction and activation of alternative lengthening of telomeres in B-lymphocytes infected by Epstein-Barr virusOncogene 2013 32:5522-30. [Google Scholar]
[9]. Sfeir A, de Lange T, Removal of shelterin reveals the telomere end-protection problemScience 2012 336:593-97. [Google Scholar]
[10]. Cawthon RM, Telomere length measurement by a novel monochrome multiplex quantitative PCR methodNucleic Acids Res 2009 37:e21 [Google Scholar]
[11]. Majewski E, Goecke T, Meinecke P, Ectrodactyly and absence (hypoplasia) of the tibia: are there dominant and recessive types?Am J Med Genet 1996 63:185-89. [Google Scholar]
[12]. Crackower MA, Scherer SW, Rommens JM, Hui CC, Poorkaj P, Soder S, Characterization of the split hand/split foot malformation locus SHFM1 at 7q21.3-q22.1 and analysis of a candidate gene for its expression during limb developmentHum Mol Genet 1996 5:571-79. [Google Scholar]
[13]. Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Mutations in the human Sonic Hedgehog gene cause holoprosencephalyNat Genet 1996 14:357-60. [Google Scholar]
[14]. Tarchini B, Duboule D, Control of Hoxd genes’ collinearity during early limb developmentDev Cell 2006 10:93-103. [Google Scholar]
[15]. Zákány J, Kmita M, Duboule D, A dual role for Hox genes in limb anterior-posterior asymmetryScience 2004 304:1669-72. [Google Scholar]
[16]. Glanz A, Fraser FC, Spectrum of anomalies in Fanconi anaemiaJ Med Genet 1982 19:412-16. [Google Scholar]
[17]. Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD, The need for more accurate and timely diagnosis in Fanconi anaemia: a report from the International Fanconi Anaemia RegistryPediatrics 1993 91:1116-20. [Google Scholar]
[18]. Mitchell JR, Wood E, Collins K, A telomerase component is defective in the human disease dyskeratosis congenitaNature 1999 402:551-55. [Google Scholar]
[19]. Armanios M, Blackburn EH, The telomere syndromesNat Rev Genet 2012 13:693-704. [Google Scholar]
[20]. Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNALancet (London, England) 2003 362:1628-30. [Google Scholar]
[21]. Dokal I, Dyskeratosis congenitaHematology Am Soc Hematol Educ Program 2011 2011:480-86. [Google Scholar]
[22]. Calado RT, Brudno J, Mehta P, Kovacs JJ, Wu C, Zago MA, Constitutional telomerase mutations are genetic risk factors for cirrhosisHepatology 2011 53:1600-07. [Google Scholar]
[23]. Holohan B, Wright WE, Shay JW, Cell biology of disease: Telomeropathies: an emerging spectrum disorderJ Cell Biol 2014 205:289-99. [Google Scholar]
[24]. Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA, Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferationOncogene 1998 16:1723-30. [Google Scholar]
[25]. Saeed H, Qiu W, Li C, Flyvbjerg A, Abdallah BM, Kassem M, Telomerase activity promotes osteoblast differentiation by modulating IGF-signaling pathwayBiogerontology 2015 16:733-45. [Google Scholar]
[26]. Abdallah BM, Haack-Sørensen M, Burns JS, Elsnab B, Jakob F, Hokland P, Maintenance of differentiation potential of human bone marrow mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene despite [corrected] extensive proliferationBiochem Biophys Res Commun 2005 326:527-38. [Google Scholar]
[27]. Cervellera M, Raschella G, Santilli G, Tanno B, Ventura A, Mancini C, Direct transactivation of the anti-apoptotic gene apolipoprotein J (clusterin) by B-MYBJ Biol Chem 2000 275:21055-60. [Google Scholar]
[28]. Huang D, Wang Y, Wang L, Zhang F, Deng S, Wang R, Poly(ADP-ribose) Polymerase 1 Is Indispensable for Transforming Growth Factor-βInduced Smad3 Activation in Vascular Smooth Muscle CellPLoS One 2011 6:e27123 [Google Scholar]
[29]. Musard JF, Sallot M, Dulieu P, Fraichard A, Ordener C, Remy-Martin JP, Identification and expression of a new sulfhydryl oxidase SOx-3 during the cell cycle and the estrus cycle in uterine cellsBiochem Biophys Res Commun 2001 287:83-91. [Google Scholar]
[30]. McPhee TR, McDonald PC, Oloumi A, Dedhar S, Integrin-linked kinase regulates E-cadherin expression through PARP-1Dev Dyn 2008 237:2737-47. [Google Scholar]
[31]. Joshi A, Mahfooz S, Maurya VK, Kumar V, Basanna CS, Kaur G, PARP1 during embryo implantation and its upregulation by oestradiol in miceReproduction 2014 147:765-80. [Google Scholar]
[32]. Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, Elledge SJ, Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repairCell 2009 138:63-77. [Google Scholar]
[33]. Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/ recombination endonucleasesCell 2009 138:78-89. [Google Scholar]
[34]. Holloway JK, Mohan S, Balmus G, Sun X, Modzelewski A, Borst PL, Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesisPLoS Genet 2011 7:e1002094 [Google Scholar]