Case 1
A seven-year-old boy with fever and persisting seizures (generalized tonic-clonic seizures) on multiple anti-epileptic medications {phenytoin (6 mg/kg/day), phenobarbitone (6 mg/kg/day) and valproate (20 mg/kg/day)} was referred to our hospital. At admission, he was in generalized convulsive status epilepticus. Blood investigations were normal. The dosage of antiepileptic medications was optimized. Cerebrospinal Fluid (CSF) analysis was suggestive of viral meningoencephalitis (lymphocytic leukocytosis, mildly elevated protein, normal glucose with sterile culture) however, specific cultivable viral titers in CSF (Japanese Encephalitis, Herpes Simplex Virus, Dengue, Enterovirus) were negative. Magnetic Resonance Imaging (MRI) brain with contrast showed increased meningeal enhancement and Electroencephalogram (EEG) showed generalized epileptiform activity. He persisted in having seizures on second line Anti-Epileptics Drugs (AEDs) {valproate 40 mg/ kg/day, levetiracetam 60 mg/kg/day} and hence was started on midazolam infusion and mechanically ventilated. Thiopental was added on, following which he remained seizure free for 120 hours with EEG showing burst suppression. Thiopental was gradually tapered and stopped however he had a recurrence of seizures. He was continued on intravenous midazolam, AEDs (Phenytoin 6 mg/kg/day, phenobarbitone 6 mg/kg/day, valproate, levetiracetam, clobazam (10 mg/day) and zonisamide 400 mg/day). He required tracheostomy for prolonged ventilation and had other comorbidities like ventilator-associated pneumonia which were managed accordingly. CSF analysis for Anti-NMDAR and voltage-gated potassium channels were negative. He gradually improved, and seizures subsided with normal EEG.
At follow up he had persistent residual neurological deficits (cognition deficits and spasticity) with occasional breakthrough seizures (generalized tonic-clonic seizures). AEDs were gradually tapered and continued on levetiracetam and clobazam. He was under regular follow up for 40 months.
Case 2
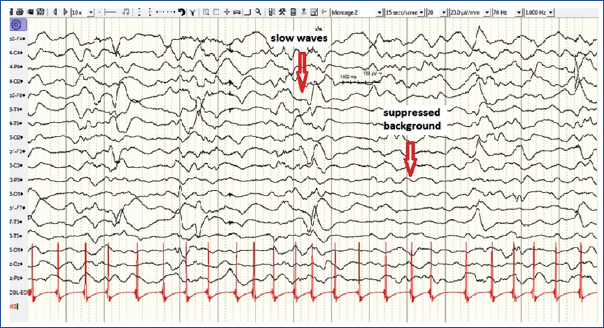
A seven-year-old girl with a history of fever and persisting seizures (generalized tonic-clonic seizures) was admitted. Blood investigations and CSF analysis including viral titers were normal. MRI brain with contrast showed subtle enhancement along the sulci. EEG showed slow waves with suppressed background [Table/Fig-1]. She had persisting seizures on phenytoin 6 mg/kg/day, phenobarbitone 8 mg/ kg/day, valproate 40 mg/kg/day, levetiracetam 60 mg/kg/day and midazolam infusion. She was intubated and mechanically ventilated, and added on ketamine (10 mg/kg/hr) infusion. She showed an inadequate response, with repeat EEG showing epileptiform activity and hence, was empirically added on pyridoxine and magnesium sulfate. Tracheostomy was done to facilitate prolonged ventilation
Interictal EEG showing slow waves with suppressed background.
She was started on methyl prednisolone suspecting autoimmune encephalitis following which seizure frequency gradually decreased, however, CSF analysis for N-methyl-D-aspartate Receptor (NMDAR) antibodies and antibodies against voltage-gated potassium channels were negative. Response to ketogenic diet was also inadequate.
Phenytoin was tapered and she was continued on phenobarbitone (6 mg/kg/day), levetiracetam (60 mg/kg/day), topiramate (15 mg/ kg/day). She sustained residual neurological deficits (variable intermittent seizures (generalized tonic-clonic, focal seizures), spastic quadriparesis, dystonia and choreo-athetoid movements) with significant morbidity (stridor, recurrent wheezing and was dependent on orogastric tube feeding) requiring frequent hospital admissions over a follow up period of 26 months.
Case 3
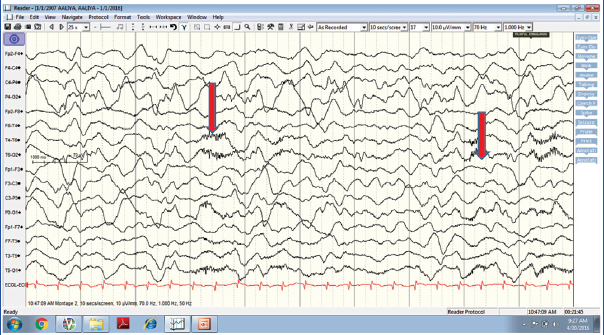
A six-year-old girl with sudden onset of unprovoked generalized tonic-clonic seizures was admitted. She had persisting seizures on benzodiazepines and was added on phenytoin, levetiracetam, valproate, lacosamide and clobazam. She had altered level of consciousness, cognitive dysfunction, mutism and orokinetic dyskinesia. Blood investigations, CSF analysis including viral studies and MRI brain were essentially normal. She was started on midazolam infusion, however, no significant response was observed. EEG showed delta brush pattern [Table/Fig-2]. Considering the clinical details and EEG findings, a diagnosis of autoimmune encephalitis was considered, and started on methylprednisolone and followed up later with Intravenous Immunoglobulin (IVIg).
EEG showing delta brush pattern.
She improved gradually, and seizures subsided, with EEG showing normalized electrical activity. CSF analysis for NMDAR antibodies and antibodies against voltage-gated potassium channels were negative. Two cycles of rituximab were given following which steroids were slowly tapered to a small daily maintenance dose (10 mg/day). Sodium valproate and lacosamide were tapered off, and levetiracetam, clobazam, phenytoin and phenobarbitone were continued. At follow up, 10 months after the onset of initial symptoms, she had occasional seizures (focal seizures + generalized tonic-clonic seizures), and cognitive deficits whereas other domains of development were normal. She was under regular follow up for 18 months.
Discussion
SRSE is defined as a status epilepticus which has persisted or recurred inspite of therapy with general anaesthesia, for 24 hours or more after the onset of treatment with anaesthesia. It includes cases where status epilepticus recurs on reduction or withdrawal of anaesthesia [1]. Though infrequently seen, it constitutes a medical emergency due to the associated high morbidity and mortality. No clear-cut guidelines are published till date for the management of super refractory status epilepticus. Most of the published literature was case reports and expert opinion [1]. We hereby reported three cases of super refractory seizures managed at a paediatric tertiary care center in coastal Karnataka, India, as they posed a therapeutic challenge.
All three cases were aged 6-7 years with prior normal developmental history and no medical illness. Blood parameters were normal in all cases.
Aetiology in the first case was probably viral meningoencephalitis (though specific viral studies were negative).
Acute encephalopathy with inflammation-mediated status epilepticus (probably Fever Induced Refractory Epileptic Encephalopathy in School-aged children-FIRES) was the probable aetiology in the second case owing to seizures with fever in the acute phase and intractable seizures with severe motor and cognitive abnormalities at a later stage. Though seizures in FIRES are known to show a better response to ketogenic diet, in this case, response to ketogenic diet was inadequate [2].
In third case, clinical features and EEG were supportive of a probable Anti-NMDA receptor encephalitis however, CSF analysis was negative for NMDAR antibodies (absence of antibodies does not exclude the possibility of immune-mediated disorder) [3].
Benzodiazepines, phenobarbitone, and phenytoin were the first line AEDs used. Levetiracetam, sodium valproate, lacosamide, clobazam, and zonisamide were used as second line agents if the response to first-line drugs was inadequate.
Midazolam infusion was used in all three cases, whereas thiopentone and ketamine in one each. Thiopental anaesthesia could achieve transient seizure control, but anaesthetic medications in other two cases could not abort the seizures. Thiopental, propofol, and midazolam were reported to attain initial control of seizures in 64%, 68%, and 78% respectively. The frequency of withdrawal seizures was highest with thiopentone (9%) whereas it was 6% and 0.3% with propofol and midazolam respectively [4].
Duration of follow up was 40 months, 26 months, and 18 months for first, second and third case respectively.
Appropriate treatment should be undertaken on an emergency basis to prevent significant brain damage due to status epilepticus. Every attempt to identify the cause of status epilepticus should be made, as it helps in providing specific treatment against the aetiology [1].
Benzodiazepines like midazolam, lorazepam or diazepam, are the initial treatment of choice and preferably should be administered as an adequate full dose rather than broken into multiple doses. A repeat dose of IV lorazepam and IV diazepam can be administered if required. This initial therapy lasts for 5-20 minutes. The second phase lasts from 20-40 minutes. IV fosphenytoin, valproic acid, levetiracetam, IV phenobarbitone are the available options in this phase. Third therapy phase starts when the seizure duration exceeds 40 minutes. Treatment options include repeating second line therapy or adding on anaesthetic agents [5].
Continuing general anaesthesia remains the mainstay treatment for super refractory status epilepticus. Concurrent continuous EEG monitoring for the first 48 hours to check for non-convulsive seizures is recommended [6]. It may not be possible in limited resource setting where intermittent EEG recordings at least 1-2 times a day would help in detecting non-clinical seizures. After an initial period of 24 hours attempt to withdraw the anaesthetic agent should be made slowly. In case of seizure recurrence, re-induction with the same anaesthetic agent and a try to withdraw the agent should be done slowly. This cyclic pattern is continued for 24-48 hours initially and over a period duration of the cycle can be increased to five days [7].
Choice of an anaesthetic agent should be individualized. Thiopentone, midazolam, and propofol are the conventional agents used. All of these agents act to enhance the action of GABA (A) receptors and thereby inducing seizure suppression. Thiopental has good anti-epileptic activity and neuroprotective action. However, because of zero order kinetics and rapid redistribution, there is an increased tendency to have a delayed recovery. At high doses, it can lead to hypotension and cardio respiratory depression. Midazolam too is known for its anti-epileptic action with a facility of rapid reversal due to its short half-life. Side-effects like development of tolerance and thereby increased risk of breakthrough seizures, cardiorespiratory depression, and hypotension are commonly seen [1,7].
Propofol is a widely used anaesthetic agent with rapid induction and withdrawal. One of the primary concern in its use is the risk of propofol infusion syndrome, especially when used at higher doses for a prolonged duration (>48 hours) and in combination either with catecholamines, steroids or ketogenic diet [1,7-9].
Ketamine is reported to be effective in few studies [1,10]. It acts by inhibiting N-methyl d-aspartate receptors and hence can be used as an alternative agent when common anaesthetic agents fail to abort the status epilepticus. It also lacks cardiorespiratory depressive action and is known to exert neuroprotective effects. Few case reports using inhalational anaesthetics like isoflurane are reported. Though the lack of ease of use and higher complication rates limit its usage, these can be useful when other treatment modalities have failed [1,11,12].
AEDs, usually no more than two or three drugs, at permitted maximal dosage are recommended [1,7]. Ideally, medications acting by same mechanisms should not be used together [13]. Drugs with least interactions and toxic effects are preferred [1,7]. [Table/Fig-3] provides the list of drugs and their dosages used in the management of super refractory status epilepticus. Frequent changes in the medications should be avoided for fear of precipitating seizures on rapid withdrawal [14-18].
Drugs and their dosages used in the management of super refractory status epilepticus.
| Drugs | Dosage and Route | Maximum Dose | Available Formulation |
|---|
| Midazolam [4,14,15] | 0.2 mg/kg, IV Infusion: 1 mcg/kg/min, increasing by 1 mcg/kg/min every 5 min till effect or max dose | Bolus: 5 mg Infusion: 12-30 mcg/kg/min have been reported [4,16]. | 1 mg/1 ml 5 ml vial |
| Lorazepam [14,15] | 0.1 mg/kg, IV | 4 mg [14,15] | 2 mg/1 ml, 2 ml amp |
| Diazepam [14] | 0.2-0.3 mg/kg, IV | 10 mg [14] | 1 ml/5 mg, 2 ml amp. |
| Phenytoin [14,15] | Loading: 18-20 mg/kg not exceeding 1 mg/kg/min to a maximum of 50 mg/min. Maintenance:4-7.5 mg/kg/day | 7.5 mg/kg/day or 300 mg/day in children [15] | 50 mg/1 ml, 2 ml amp |
| Fosphenytoin [15,17,18] | 20 phenytoin equivalents/kg, at 3 PE/kg/min. maintenance dose: 4-5 mg/kg/day, at an infusion rate of 1-2 mg/kg/min, not exceeding 100 mg/min | Maximum infusion rate for loading dose 150 mg/min [15] | 75 mg/1 ml, 10 ml vial |
| Phenobarbitone [14-16] | Loading: 15-20 mg/kg, infusion not exceeding 1 mg/kg/min to a maximum of 1000 mg [15]. In adults, rate should not exceed 100 mg/min. | Maintenance of 2.5-5 mg/kg/day till a maximum dose of 600 mg/ day in two divided doses [15]. | 1 ml/200 mg, 1 ml amp |
| Valproate [15,16] | Loading:10-20 mg/kg infusion intravenously not exceeding 6 mg/kg/min Continuous infusion of 1-2 mg/ kg/hr. Maintenance dose 10-20 mg/kg/day. | 35-40 mg/kg/day to a maximum of 2500 mg/day.[15] | 1 ml/100 mg, 5 ml vial |
| Levetiracetam [14,16] | Loading: 20 mg/kg, maintenance: upto 60 mg/ kg/day | 3000 mg/day [15] | 1 ml/100 mg, 5 ml vial |
| Topiramate [14,16,21] | Initial 5-10 mg/kg/day, PO for two days, followed by 5 mg/ kg/day/in two divided doses as maintenance. The dose can be increased gradually once in one to two weeks. | Adjunctive therapy: upto15- 25 mg/kg/ day in few studies. Monotherapy: upto 16 mg/ kg/day, to a maximum of 400 mg/day [15] | 25 mg/50 mg Tablet |
| Lidocaine [14] | 2 mg/kg IV bolus not exceeding 50 mg/min, followed by a maintenance of 2 mg/kg/hr | - | 20 mg/1 ml (5 ml vial) 10 mg/1 ml (20 ml vial) |
| Lacosamide [18] | Lacosamide: 4-12 mg/kg/day. | Upto 20 mg/kg/ day, to a maximal dose of 600 mg/ day. [18] | 10 mg/1 ml, 20 ml vial |
| Clobazam [15] | one month-12 years: 0.125 mg/ kg twice daily till maximum of 0.5 mg/kg/day. 12-18 years: 10 mg/twice daily. | 1month-12 years: 15 mg twice daily. 12-18 years: 30 mg twice daily [15]. | |
| Thiopental [1,4,14] | Induction: 5 mg/kg, followed by maintenance 5 mg/kg/hr, to increase every 2 min by 1 mg/ kg/hr till burst suppression occurs. Infusion to continue for 12-48 hrs. | 12 mg/kg/hr [1] | 500 mg, 1000 mg vial |
| Propofol [1] | Loading:1-2 mg/kg Maintenance: 1-7 mg/kg/hr | 26.94 mg/kg/ hr [1] | 10 mg/ml, 50 ml vial |
| Ketamine [4] | 1-3 mg/kg/hr | Upto 5 mg/kg/ hr [4] | 50 mg/1 ml, 5 ml vial |
| Magnesium sulphate [1,4] | Bolus: 4000 mg Infusion: 2000-6000 mg/hr, to maintain serum level to 3.5 mmol/L | 6 gm/hr [1] | 50% w/v 2 ml amp |
| Pyridoxine [1,4,15] | 30 mg/kg in children [1] | 300 mg [1] | 40/100 mg tablet |
| Steroids [1] | Methyl prednisolone 1 gram/ day, intravenous for 3 days followed by 1 mg/kg/day. | 1000 mg/day [1,7] | 500 mg, 1000 mg/vial |
| IVIg [1,4] | 400 mg/kg/day for 5 days | 2 gm/kg [1,4] | 5000 mg/100 ml |
IV: intravenous, IVIg: Intravenous Immunoglobulin, PO: per oral, mg: milligram, ml: milliliters.
The recent discovery of the loss of GABA receptor density due to receptor trafficking [19-22] during status epilepticus is considered as one of the mechanisms leading to the failure of GABAergic drugs like benzodiazepines and phenobarbitone. This highlights the need for the use of medications which have different mechanisms of action to control seizures. Allopregnanolone a neurosteroid acts as a positive allosteric modulator of GABA (A) receptors (synaptic and extrasynaptic) and thereby may overcome resistance to benzodiazepines and barbiturates [23].
Intravenous magnesium sulfate infusion, though reported to be effective in few studies [24], lacks convincing evidence to recommend as a treatment modality. Pyridoxine/pyridoxal phosphate is curative in children with a mutation in ALDH7A1 gene (encoding antiquitin), presenting with status epilepticus [1,23].
Association of antibodies against receptors like voltage-gated potassium channels and N-methyl d-aspartate receptors in cryptogenic status epilepticus as well as evidence of the role of inflammation [25] in the initiation of epilepsy have led to the use of immunotherapy in cases failing to respond to conventional treatment. Corticosteroids, IVIg and plasma exchange, have been used as a part of immunotherapy. Immunomodulatory therapy is useful in Rasmussen encephalitis, Hashimoto’s encephalitis and vasculitis [21]. Recently there is a surge in use of immunotherapy considering immune mechanism as a probable underlying cause in cases where precise aetiology could not be identified [1,7].
Ketogenic diet by its anti-inflammatory action has been successful, in few studies [25,26] in controlling super refractory status epilepticus. It is useful in children with GLUT-1 transporter deficiency and pyruvate carboxylase deficiency [23].
Therapeutic hypothermia by reducing the cerebral metabolic rate and by decreasing cerebral oedema, free radical production, and oxidative stress has been used as an additive measure in the treatment of super refractory seizures. Surgical resection of a clearly defined epileptiform focus can be attempted if status epilepticus persists inspite of appropriate medical management. Other modes of intervention include vagal nerve stimulation, transcranial magnetic stimulation, electroconvulsive therapy and cerebrospinal fluid drainage for persisting status epilepticus [1].
Conclusion
Super refractory status epilepticus is a medical emergency neces sitating aggressive treatment for early control seizures. It is associated with high rates of mortality and morbidity (associated serious brain damage, respiratory infections, feeding problems). Aetiology may not be identified in few cases inspite of the extensive workup, further aggravating the challenges in management. An integrated and comprehensive intensive care would help in managing this challenging condition.
IV: intravenous, IVIg: Intravenous Immunoglobulin, PO: per oral, mg: milligram, ml: milliliters.
[1]. Shorvon S, Ferlisi M, The treatment of super-refractory status epilepticus: A critical review of available therapies and a clinical treatment protocolBrain 2011 134:2802-18. [Google Scholar]
[2]. Nabbout R, Vezzani A, Dulac O, Chiron C, Acute encephalopathy with inflammation-mediated status epilepticusThe Lancet Neurology 2011 10:99-108. [Google Scholar]
[3]. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, A clinical approach to diagnosis of autoimmune encephalitisLancet Neurol 2016 15(4):391-404. [Google Scholar]
[4]. Shorvon S, Ferlisi M, The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapyBrain 2012 135(8):2314-28. [Google Scholar]
[5]. Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American epilepsy societyEpilepsy Curr 2016 16(1):48-61. [Google Scholar]
[6]. Brophy GM, Bell R, Claassen J, Alldredge B, Bleck TP, Glauser T, Guidelines for the evaluation and management of status epilepticusNeurocrit Care 2012 17(1):3-23. [Google Scholar]
[7]. Shorvon S, Super-refractory status epilepticus: an approach to therapy in this difficult clinical situationEpilepsia 2011 52(8):53-56. [Google Scholar]
[8]. Power KN, Flaatten H, Gilhus NE, Engelsen BA, Propofol treatment in adult refractory status epilepticus. Mortality risk and outcomeEpilepsy Res 2011 94(1–2):53-60. [Google Scholar]
[9]. Iyer VN, Hoel R, Rabinstein A, Propofol infusion syndrome in patients with refractory status epilepticus: an 11-year clinical experienceCrit Care Med 2009 37(12):3024-30. [Google Scholar]
[10]. Borris DJ, Bertram EH, Kapur J, Ketamine controls prolonged status epilepticusEpilepsy Res 2000 42(2–3):117-22. [Google Scholar]
[11]. Zhumadilov A, Gilman CP, Viderman D, Management of super-refractory status epilepticus with isoflurane and hypothermiaFront Neurol 2015 6:286 [Google Scholar]
[12]. Mirsattari SM, Sharpe MD, Young GB, Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desfluraneArch Neurol 2004 61(8):1254-59. [Google Scholar]
[13]. Lee JW, Dworetzky B, Rational polytherapy with antiepileptic drugsPharmaceuticals 2010 3:2362-79. [Google Scholar]
[14]. Capovilla G, Beccaria F, Beghi E, Minicucci F, Sartori S, Vecchi M, Treatment of convulsive status epilepticus in childhood: Recommendations of the Italian League Against EpilepsyEpilepsia 2013 54:23-34. [Google Scholar]
[15]. Sweetman S, Martindale 2009 36th edChicagoPharmaceutical Press:477-513.986-1005, 1978-80 [Google Scholar]
[16]. Mishra D, Sharma S, Sankhyan N, Konkani R, kamate M, Kanhere S, Consensus guidelines on management of childhood convulsive status epilepticusIndian Pediatrics 2014 51:975-90. [Google Scholar]
[17]. Broomall E, Natale JE, Grimason M, Goldstein J, Smith CM, Chang C, Pediatric super-refractory status epilepticus treated with allopregnanoloneAnn Neurol 2014 76(6):911-15. [Google Scholar]
[18]. Buck ML, Goodkin HP, Use of lacosamide in children with refractory epilepsyJ Pediatr Pharmacol Ther 2012 17(3):211-19. [Google Scholar]
[19]. Arancibia-Carcamo IL, Kittler JT, Regulation of GABA(A) receptor membrane trafficking and synaptic localizationPharmacol Ther 2009 123(1):17-31. [Google Scholar]
[20]. Smith KR, Kittler JT, The cell biology of synaptic inhibition in health and diseaseCurrent Opinion in Neurobiology 2010 20:550-56. [Google Scholar]
[21]. Abend NS, Loddenkemper T, Management of pediatric status epilepticusCurr Treat Options Neurol 2014 16(7):301 [Google Scholar]
[22]. Naylor DE, Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticusJ Neurosci 2005 25(34):7724-33. [Google Scholar]
[23]. Broomall E, Natale JE, Grimason M, Goldstein J, Smith CM, Chang C, Pediatric super-refractory status epilepticus treated with allopregnanoloneAnn Neurol 2014 76(6):911-15. [Google Scholar]
[24]. Visser NA, Braun KPJ, Leijten FSS, Van Nieuwenhuizen O, Wokke JHJ, Van Den Bergh WM, Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutationsJ Neurol 2011 258(2):218-22. [Google Scholar]
[25]. Vezzani A, Rüegg S, Immunity and inflammation in EpilepsyIntroduction Epilepsia 2011 52:1-4. [Google Scholar]
[26]. Kumada T, Miyajima T, Kimura N, Saito K, Shimomura H, Oda N, Modified Atkins diet for the treatment of nonconvulsive status epilepticus in childrenJ Child Neurol 2010 25(4):485-89. [Google Scholar]
[27]. Nabbout R, Mazzuca M, Hubert P, Peudennier S, Allaire C, Flurin V, Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES)Epilepsia 2010 51(10):2033-37. [Google Scholar]