Adult Survival in Ellis-van Creveld Syndrome with Common Atrium – A Rare Case Report
Mugula Sudhakar Rao1, Devavrata Sahu2, Hashir Kareem3, Tom Devasia4, Kishor Kumar Shetty5
1 Registrar, Department of Cardiology, KMC, Manipal University, Manipal, Karnataka, India.
2 Junior Resident, Department of Medicine, KMC, Manipal University, Manipal, Karnataka, India.
3 Associate Professor, Department of Cardiology, KMC, Manipal University, Manipal, Karnataka, India.
4 Professor, Department of Cardiology, KMC, Manipal University, Manipal, Karnataka, India.
5 Consultant Physician, Department of Medicine, Vinaya Hospital, Kundapur, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Hashir Kareem, Associate Professor, Department of Cardiology, KMC, Manipal University, Manipal-576104, Karnataka, India.
E-mail: hashirkareem@gmail.com
Ellis-van Creveld syndrome is a rare genetic disorder, characterized by chondral dysplasia, ectodermal dysplasia, polydactyly and congenital heart defects. Patients with this syndrome rarely survive into adulthood. The syndrome has also been rarely reported in the Indian population. We present the case of a 56 year old female who presented with post-menopausal bleeding, and was diagnosed with Ellis van Creveld syndrome, with a common atrium and common atrioventricular valve.
Genetic disorder, Polydactyly, Post-menopausal bleeding
Case Report
A 56-year-old female, with no known pre-morbidities, was admitted for evaluation of postmenopausal bleeding. Patient had attained menopause at the age of 50 years, and she had recently noticed spotting over 10-15 days, prior to the admission. On general examination, patient was short statured, with a height of 120 centimeters. Upper and lower body segments were disproportionate and were in a ratio of 1:1 as compared to the normal adult ratio of 0.9. Multiple oral and bony deformities were noted. Patient had an abnormal dentition, with small poorly formed teeth. Gingival sulcus was narrow. She gave history of having used a set of dentures since childhood. She also gave history of having undergone minor procedures in the past for removal of 'additional adherent tags' between the lips and gums.
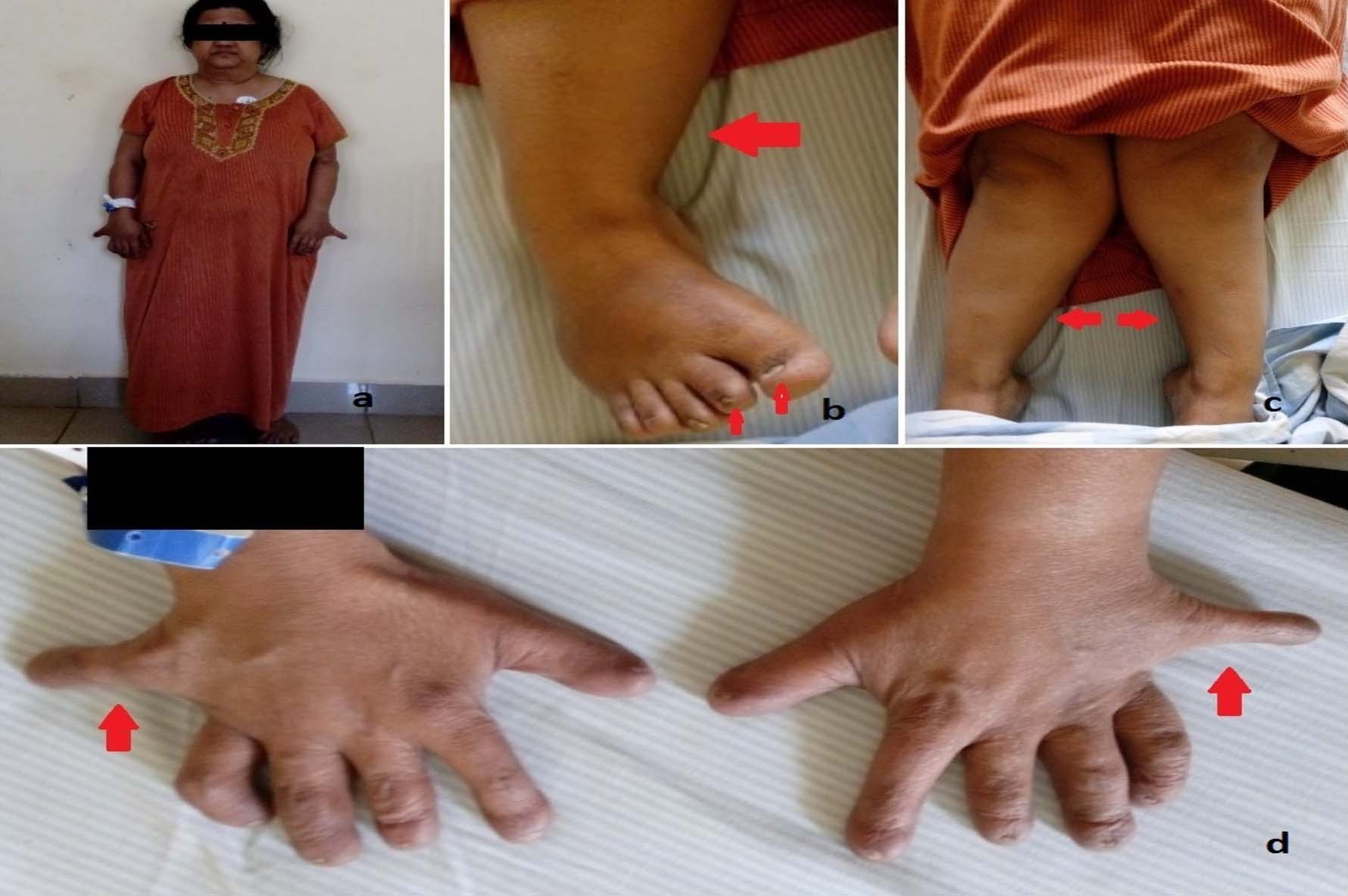
Skeletal system examination revealed forearms to be disproportionately short compared to the arms and hands bilaterally. Cubitus valgus was present. Post axial polydactyly was noted bilaterally and fingernails were poorly developed. In the lower limbs, legs were disproportionately short compared to thighs and feet bilaterally. Genu valgus was present bilaterally. Feet also had poorly developed nails. No polydactyly was noted in the lower limbs [Table/Fig-1a-d].
a) Profile of the patient showing short stature; b) Poorly developed nails in the feet (small arrows), with mesomelia of the lower limbs (large arrow); c) Genu valgum and mesomelia of the lower limbs; d) Polydactyly, clinodactyly of the fifth digits (arrows), and poorly developed nails in the hands bilaterally.
Cardiovascular examination revealed a long narrow thorax, grade 3/6 short systolic murmur at the left lower sternal border and a loud second heart sound. Rest of the cardiovascular examination was unremarkable.
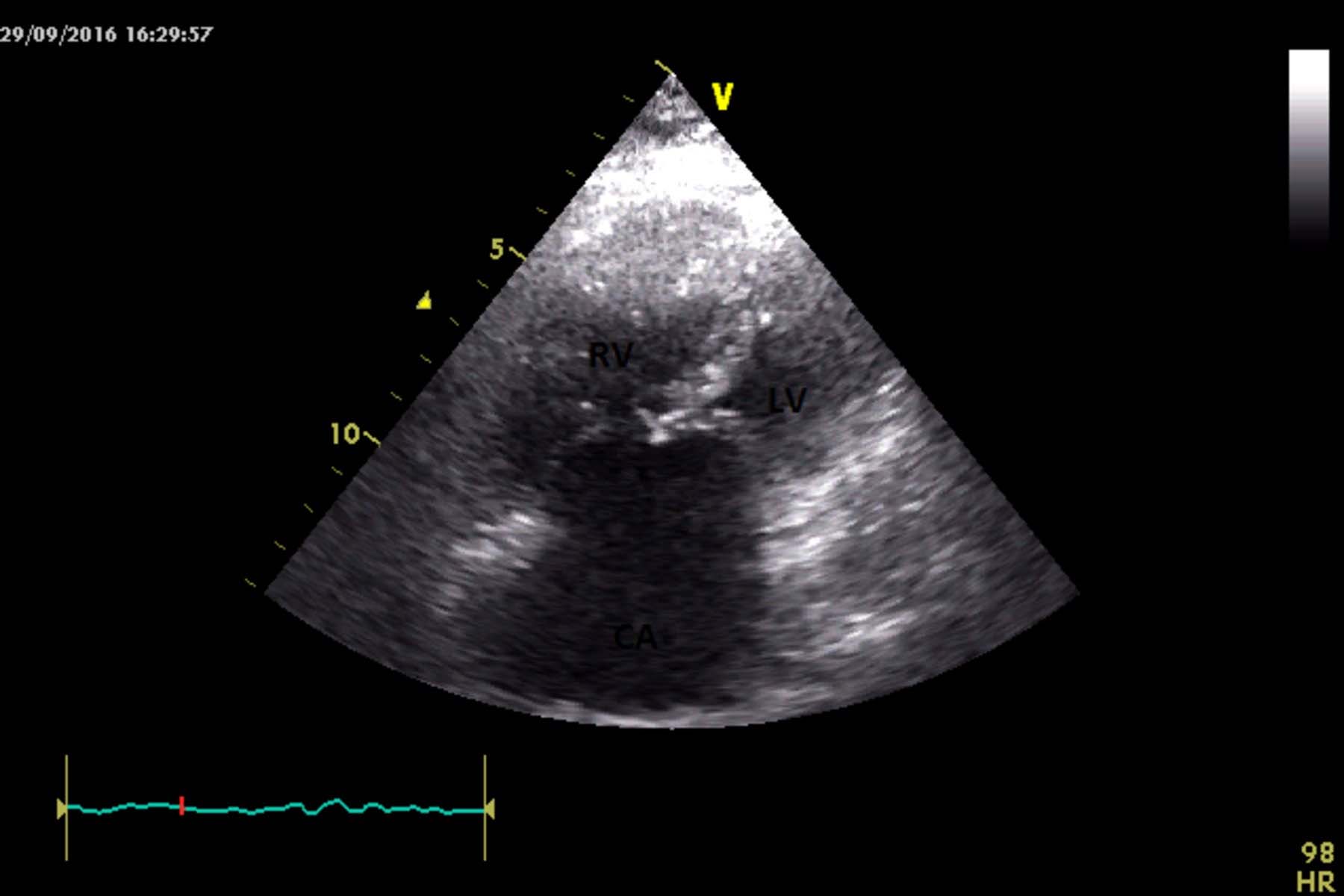
With the suspicion of a syndromic diagnosis, an echocardiogram was done which showed a common atrium, common atrioventricular valve and severe pulmonary artery hypertension [Table/Fig-2].
Two dimensional echocardiographic image of common atrium with common atrioventricular valve (apical four chamber view) showing Right Ventricular (RV) and Left Ventricular (LV) cavity with Common Atrium (CA).
Patient had been born of a non-consanguineous marriage. No other family member had similar features. There was no family history of neonatal or childhood deaths to the best of their knowledge.
In the light of the aforementioned features, a clinical diagnosis of Ellis-van Creveld syndrome was made.
Gynaecological evaluation was performed in the form of colposcopy with cervical biopsy, and fractional curettage. Reports were negative for malignancy, and only showed squamous metaplasia with inflammatory atypia on the cervical biopsy. Patient was on regular follow up for the same. Family members were counselled regarding the genetic basis of her presentation, and the need for further assessment by genetics for possible manifestations in future generations.
Discussion
Ellis-van Creveld syndrome is a rare, autosomal recessive genetic disorder characterized by tetrad of chondral dysplasia, ectodermal dysplasia, polydactyly and congenital heart defects [1].
Mutations in EVC1 and EVC2 genes, located in head to head configuration on chromosome 4 (4p16), are responsible for the clinical manifestations [1]. Truncating mutations, amino acid deletions and missense mutations have all been described as causal. Since it is an autosomal recessive disorder, there is one in four chance of the person having the clinical syndrome [2]. Studies are in progress to assess expression of the phenotype in individuals heterozygous for the genetic defects [1]. Studies have also reported biological closeness of parents as having a direct impact on the severity of manifestations; closer the parents biologically, more severe the manifestations. The disease has been most commonly reported in the Amish community of Pennsylvania, USA, and Ashkenazi Jews [1].
Manifestations of the disorder appear to involve all embryonic layers [3]. Ectodermal dysplasia is characterized by abnormal hair growth, abnormal nail formation and poor dentition. Enamel hypoplasia, hypodentia, narrow gingival sulcus, presence of accessory frenulae are some of the oral manifestations of EVC [1,2]. Mesodermal involvement primarily affects skeletal and cardiac development. Chondral dysplasia takes the form of acromesomelia, with progressive shortening of the distal extremities, along with associated polydactyly. Other skeletal defects include cubitus valgus and genu valgus. Radiologically, supernumary carpals, fused carpals, abnormal bone maturation, lag of bone age and tubular bones can be seen [2]. A multitude of cardiac defects has been described; of note is the presence of a common atrium due to agenesis of atrial septum [3,4]. Although an uncommon congenital defect by itself, it occurs at a high rate in EVC. Our patient had cubitus valgus, genu valgum and post axial polydactyly. Evidence of ectodermal dysplasia was present in the form of poor dentition and poorly developed nails. Cardiac abnormality was manifested in the form of common atrium.
A retrospective review of data regarding congenital heart disease in EVC between 1982-2007, by Hills CB et al., found that of 32 paediatric patients, 28 had an endocardial cushion defect, with 15 patients having a common atrium [4]. ASD, VSD, hypoplastic left heart and other valve defects have also been described in relation with EVC. Liver and lung pathologies have been reported but primary endodermal involvement is rare in EVC [5]. Epispasdias, hypospasdias and cryptorchidism have also been reported to have some association with EVC [1].
Review of literature shows no association between EVC and post-menopausal bleeding. Da Silva EO et al., described 15 cases of EVC in an inbred family, with the eldest survivor being 82 years old [6]. Review of literature also gives sporadic instances of patients surviving up to 35-40 years of age [7]. However, such cases have rarely been reported from India [8,9], further highlighting its exceptional nature. To the best of our knowledge, it is one of the rare case reports of EVC surviving into adulthood, from India.
Conclusion
With this case report, we emphasize the fact that in any patient presenting with cardiovascular anomaly and dysmorphic features, a syndromic diagnosis should always be considered. We also like to highlight the rare occurrence of this disorder and even rarer survival to adulthood.
[1]. Baujat G, Le Merrer M, Ellis-van Creveld syndromeOrphanet J Rare Dis 2007 Jun 4 2:27 [Google Scholar]
[2]. Cesur Y, Yuca SA, Üner A, Yuca K, Arslan D, Ellis-Van Creveld SyndromeEur J Gen Med 2008 5(3):187-190. [Google Scholar]
[3]. Francis L, Giknis Single atrium and the Ellis—van Creveld syndromeThe Journal of Pediatrics62(4):558-64. [Google Scholar]
[4]. Hills CB, Kochilas L, Schimmenti LA, Moller JH, Ellis-van Creveld syndrome and congenital heart defects: presentation of an additional 32 casesPediatr Cardiol 2011 Oct 32(7):977-82. [Google Scholar]
[5]. Hegde K, Puthran RM, Nair G, Nair PP, Ellis van Creveld syndrome-a report of two siblingsBMJ Case Rep 2011 [Google Scholar]
[6]. da Silva EO, Janovitz D, de Albuquerque SC, Ellis-van Creveld syndrome: report of 15 cases in an inbred kindredJ Med Genet 1980 Oct 17(5):349-56. [Google Scholar]
[7]. Ray HN, “A Case Report of Late Survival in Ellis: Van Creveld Syndrome”Journal of Evolution of Medical and Dental Sciences 2014 3(55):12690-12694. [Google Scholar]
[8]. Mishra T, Routray SN, Das B, Late survival in Ellis-van Creveld syndrome – a case reportIndian Heart J 2012 64(4):408-11. [Google Scholar]
[9]. Kumar A, Sharma S, Sharma M, Bhargava K, Common atrium with unusual electrocardiogram in Ellis-van Creveld syndrome: A case reportHeart India 2015 3:103-05. [Google Scholar]