Juvenile Hyaline Fibromatosis- A Rare Autosomal Recessive Disease
Prarthana Sameer Kalgaonkar1, Minal Wade2, Charusheela Warke3, Meena Makhecha4, Manisha Khare5
1 Consultant, Department of Paediatrics, Dr R N Cooper Hospital and Hindu Hriday Samrat Balasaheb Thackrey Medical College, Vile Parle West, Mumbai, India.
2 Associate Professor, Department of Paediatrics, Dr R N Cooper Hospital and Hindu Hriday Samrat Balasaheb Thackrey Medical College, Vile Parle West, Mumbai, India.
3 Associate Professor, Department of Paediatrics, Dr R N Cooper Hospital and Hindu Hriday Samrat Balasaheb Thackrey Medical College, Vile Parle West, Mumbai, India.
4 Associate Professor, Department of Dermatology, Dr R N Cooper Hospital and Hindu Hriday Samrat Balasaheb Thackrey Medical College, Vile Parle West, Mumbai, India.
5 Professor, Department of Pathology, Dr R N Cooper Hospital and Hindu Hriday Samrat Balasaheb Thackrey Medical College, Vile Parle West, Mumbai, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Prarthana Sameer Kalgaonkar, E-10, Brindavan Society, Koldongri Sahar Road, Andheri East Mumbai-400069, India.
E-mail: doctorprarthana@yahoo.com
Systemic hyalinosis is inherited as an autosomal recessive disease. It may also be referred to as Fibromatosis hyalinica multiplex juvenilis and Murray-Puretic-Drescher syndrome. A four and a half-year-old female child presented with multiple soft tissue swellings involving the nose, orbital ridges, ears, bony prominences of the ulna and tibia and the parietal and occipital prominence and had gum hypertrophy. The diagnosis of this rare condition was based upon clinicopathological correlation, wherein the histopathological examination of cutaneous lesions reveals accumulation of hyaline material with fibroblast in the dermis. A multidisciplinary approach helped in correct diagnosis, management and in providing counseling for the parents. The child’s parents were counseled about the surgical excision of the lesion; however, the parents opted for non-surgical conservative management.
Gingival hyperplasia, Hyaline deposition, Nodular skin lesions
Case Report
A four and a half-year-old girl presented to paediatric department with multiple soft tissue swellings affecting the ears, nose, the upper back and scalp along with firm swellings over the long bones [Table/Fig-1].
Soft tissue swelling involving ears, nose and the nasolabial fold, and bony swelling on right orbital ridge.
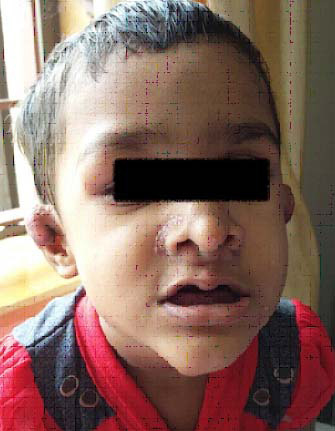
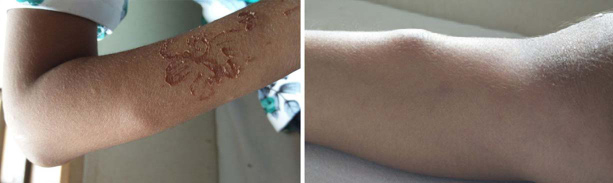
The child was first in order of birth, born of a non- consanguineous marriage. Birth history was uneventful. She started developing swellings over the ears, nose since the age of three years. These swellings were multiple, firm, non-tender, waxy papules and nodules involving the tip of nose, nasolabial folds and ears, which were small to begin with and progressively increased in size. By four years of age she also developed three, soft, skin-coloured, non-tender, pleomorphic masses, measuring up to 3 cm in diameter, over the parietal and occipital eminences of the scalp. Simultaneously she developed a firm swelling along the superior orbital ridge, followed by firm non-tender soft tissue swellings over the bony prominences along the ulnar aspect of the forearm and the tibia bilaterally [Table/Fig-2a,b]. There was no joint restriction.
a) Firm and non tender swelling over the ulnar aspect of the forearm; b) firm non-tender swelling over the tibia.
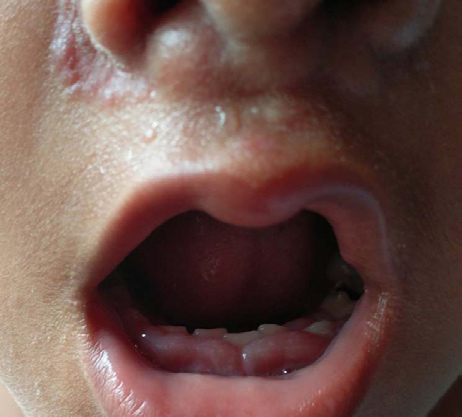
When she presented with these complaints, her vitals were stable. There was no lymphadenopathy or hepatosplenomegaly. The head circumference was 52 cm whereas her weight and height was 11 kg (<3rd percentile) and 91 cm (<3rd percentile) respectively, implying severe acute malnutrition. She had normal motor and developmental milestones. Her dentition correlated with her age while on examination there was gum hypertrophy [Table/Fig-3]. She had normal hearing and vision. There was no other systemic involvement.
The complete haemogram, renal function tests, liver function tests, electrolytes and ultrasound of the abdomen were performed to rule out systemic involvement. These were within normal limits. Radiograph of the chest and the skeletal survey were normal. Electrocardiography and 2D-echocardiography did not reveal any cardiac abnormality.
At this point of time, differential diagnosis included neurofibromatosis, juvenile hyaline fibromatosis, congenital generalized fibromatosis, Winchester syndrome.
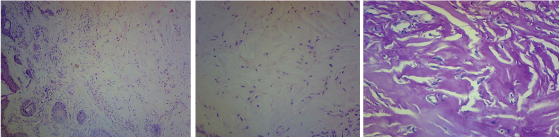
Considering the cutaneous involvement, a skin biopsy was performed from ala nasi. The histopathological reports of skin biopsy showed an ill circumscribed dermal nodule composed of spindle shaped cells embedded in a homogeneous eosinophilic matrix with fibroblast within {[Table/Fig-4a] low power and [Table/Fig-4b] high power}. This homogeneous eosinophilic matrix was PAS positive [Table/Fig-4c]. No elastic tissue was seen. These deposits were Congo red-negative and Alcian blue-negative.
The skin biopsy showed; a) H/E stain (10X) low power - Dermis, poorly circumscribed hypocellular lesion composed of cytologically bland cells embedded in an abundant hyalinized stroma; b) H/E stain (40X). High power –homogeneous eosinophilic matrix with spindle cells. c) PAS staining of the tissue, there is absence of elastic tissue and abundant hyaline material in the backdrop (40X)
The diagnosis of juvenile hyaline fibromatosis was made after clinicopathological correlation. The parents were counseled about the progressive nature of the disease and the 25% chance of an affected future offspring [1]. We could not go ahead with the genetic testing in our patient due to financial constraints.
The parents were offered surgical excision of the facial lesions for cosmetic purpose however in view of high rate of recurrence they deferred surgical management and opted for conservative treatment. There was no joint restriction or problems related to gum hypertrophy in our case so no active management was offered. The child followed up in the outpatient department after three months of diagnosis to watch for progression of disease which may compromise the quality of life in the form of development of joint restriction and progressive gum hypertrophy that may hamper feeding. However, on follow up there was no significant increase in the size of the lesions involving the nose and the ears. Hence no specific treatment was required and was asked to follow up every three monthly.
Discussion
Juvenile hyaline fibromatosis/inherited systemic hyalinosis is a rare, autosomal recessive and hereditary disease with distinct clinical and histopathological features. There is paucity of available literature with only < 70 cases reported worldwide and a few case reports from India [1-5].
The mutant gene that results in Inherited hyalinosis syndrome has been mapped to 4q21 [6]. Abnormal biosynthesis of glycosaminoglycans and collagen III- VI has been defined. Another study identified a mutations in the capillary morphogenesis factor-2 gene which have been shown to be pathogenetic in development of Inherited systemic hyalinosis [7]. The disease commonly affects children in the age group of 2-5 years. Symptoms usually develop in the first two years of life [3]. These include gingival hypertrophy, papules distributed around the nose, the ears, in the genital area and on the thighs. Patients develop joint contractures secondary to fibrosis and mass effect. These lesions can be large and deforming at times. Excessive skin stretching and distraction leads to cutaneous pressure necrosis and ulceration. These are prone to infective complications [3]. Joint contractures cause increasing morbidity and eventually the patients are bedridden [8,9].
Infantile systemic hyalinosis is a severe form of hyaline fibromatosis syndrome with progressive joint involvement, joint contractures and cutaneous thickening in early stage of the disease. They develop repeated purulent infections, diarrhea and osteoporosis in early stages of life followed by hyaline infiltration of the intestinal mucosa causing protein-losing enteropathy subsequent malnutrition. Severe joint limitation and pain lead to immobility and respiratory insufficiency. Sepsis, respiratory/ cardiac failure are common causes of mortality in these patients [10,11].
Juvenile hyaline fibromatosis and Infantile systemic hyalinosis, both these conditions are linked to a disorder in the synthesis of glycosaminoglycans with consequent abnormality in collagen synthesis.
Close differentials of the condition due to the cutaneous nodules were neurofibromatosis, congenital generalized fibromatosis while isolated gingival hyperplasia, Winchester syndrome, and lipoid proteinosis may also be considered as differential diagnoses [10].
Treatment is predominantly symptomatic and palliative. There is no definitive treatment available. Early surgical excision may help to prevent the appearance of new lesions [12], although recurrence has been documented after excision [8]. Intralesional steroid injection may reduce the size/volume of early lesions. Surgical excision is limited for those cases with life limiting dysmorphism or with significant functional impairment. More invasive surgical options such as partial gingivectomy may be considered in more aggressive debilitating cases. Conservative therapy may include oral D-penicillamine [1,13]. Though, the prognosis is better in infantile systemic hyalinosis, the patients are left with deformities and joint contractures.
Conclusion
Juvenile hyaline fibromatosis is a rare autosomal recessive disease. Clinical suspicion prompts histopathological examination of the lesions followed by medical and surgical intervention as a part of palliative care. A multidisciplinary approach is required in the diagnosis and management of juvenile hyaline fibromatosis and to provide adequate genetic and preconception counseling to the parents in order to prevent an affected future offspring
[1]. Krishnamurthy J, Dalal BS, Sunita MV, Gubbanna. Juvenile hyaline fibromatosisInd J Dermatol 2011 56(6):731-33. [Google Scholar]
[2]. Gupta LK, Singhi MK, Bansal M, Khullar R, Jain V, Kachhawa D, Juvenile hyaline fibromatosis in siblingsIndian J Dermatol Venereol Leprol 2005 71:115-18. [Google Scholar]
[3]. Nischal KC, Sachdev D, Kharkar V, Mahajan S, Juvenile hyaline fibromatosisJ Postgrad Med 2004 50:125-26. [Google Scholar]
[4]. Varshini KA, Haritha K, Hyaline Fibromatosis SyndromeIndian J Paediatr Dermatol 2016 17:38-41. [Google Scholar]
[5]. Rashmi MV, Geetha JP, Arava S, Murthy N, Kodandaswamy CR, Juvenile Hyaline Fibromatosis (JHF): a rare case with recurrenceJ Clin Diagn Res 2014 8(2):161-62. [Google Scholar]
[6]. Rahman N, Dustan M, Teare MD, Hanks S, Edkins SJ, Hughes J, The gene for JHF maps to 4q21Am J Hum Genet 2002 71:975-80. [Google Scholar]
[7]. Hanks S, Adams S, Douglas J, Arbour L, Atherton DJ, Balci S, Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosisAm J Hum Genet 2003 73:791-800. [Google Scholar]
[8]. Finlay AY, Ferguson SD, Holt PJ, Juvenile hyaline fibromatosisBr J Dermatol 1983 108:609-16. [Google Scholar]
[9]. Burgdorf W, Ruiz-Maldonado R, Benign and malignant tumors. In: Schachner L.A, Hansen R, editorsPaediatric Dermatology 2003 3rd edEdinburghElsevier Limited:870 [Google Scholar]
[10]. Shin HT, Paller A, Hoganson G, Willner JP, Chang MW, Orlow SJ, Infantile systemic hyalinosisJ Am Acad Dermatol 2004 50:S61-64. [Google Scholar]
[11]. Glover MT, Lake BD, Atherton DJ, Clinical, histologic and ultrastructural findings in two cases of infantile systemic hyalinosisPaediatr Dermatol 1992 9:255-58. [Google Scholar]
[12]. Huang YC, Xiao YY, Zheng YH, Jang W, Yang YL, Zhu XJ, Infantile systemic hyalinosis: A case report and mutation analysis in a Chinese infantBr J Dermatol 2007 156:602 [Google Scholar]
[13]. Urbina F, Sazunic I, Murray G, none Infantile systemic hyalinosis or juvenile hyaline fibromatosis?Paediatr Dermatol 2004 21:154-59. [Google Scholar]