A Case of Hyperargininaemia Presenting at Unusually Low Age
Vanita Lal1, Daisy Khera2, Garima Gupta3, Kuldeep Singh4, Praveen Sharma5
1 Associate Professor, Department of Biochemistry, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India.
2 Associate Professor, Department of Paediatrics, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India.
3 Senior Resident, Department of Biochemistry, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India.
4 Professor and Head, Department of Paediatrics, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India.
5 Senior Professor and Head, Department of Biochemistry, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Vanita Lal, Associate Professor, Department of Biochemistry, AIIMS, Basni Industrial Area, Phase 2, Jodhpur, Rajasthan, India.
E-mail: laldasvanita@gmail.com
Arginase or ARG1 gene deficiency is a Type V Urea Cycle Disorder (UCD) (catalysing the fifth reaction of urea cycle), associated with hyperammonaemia. Here, we discuss a rare case of a 13-month-old female, having Severe Acute Malnutrition (SAM) and failure to thrive, with serial high plasma ammonia, normal plasma lactate with high arginine and glutamine levels on Amino Acid Assay (AAA) which was performed on 1220 Agilent HPLC. She was admitted for about a month and eventually succumbed to her ailment after a month of discharge.
Arginase, Glutamate, Hyperammonaemia, Normal lactate, Orotate, Urea cycle disorder
Case Report
A 13-month-old female presented with initial complaints of not gaining weight since birth, recurrent vomiting and tonic posture till tenth day of her life. She had a past history of seizures at the age of 6-7 months associated with fever with a frequency of 1-2 episodes per month which lasted till 11 months of age. She was admitted at another tertiary care hospital at the age of 10 months for not gaining weight and was diagnosed with global developmental delay.
She was the third child of a second degree consanguineous marriage. Mother’s antenatal period was uneventful with proper intake of iron and folate supplementation. The girl was born full term by Lower Segment Caesarean Section (LSCS) in hospital. The birth weight was 1.8 kg, with immediate cry after birth. Breast feeding was started 3-4 hours after birth. The early neonatal period was uneventful. She received complete immunisation with DPT/OPV/Hep B vaccination.
On general physical examination, she was found undernourished with presence of pallor. Her height/weight ratio was less than -3 SD. A differential diagnosis of global developmental delay, sepsis, SAM, iron deficiency anaemia or inborn error of metabolism was made. Then, the child was admitted and started with nasogastric feed.
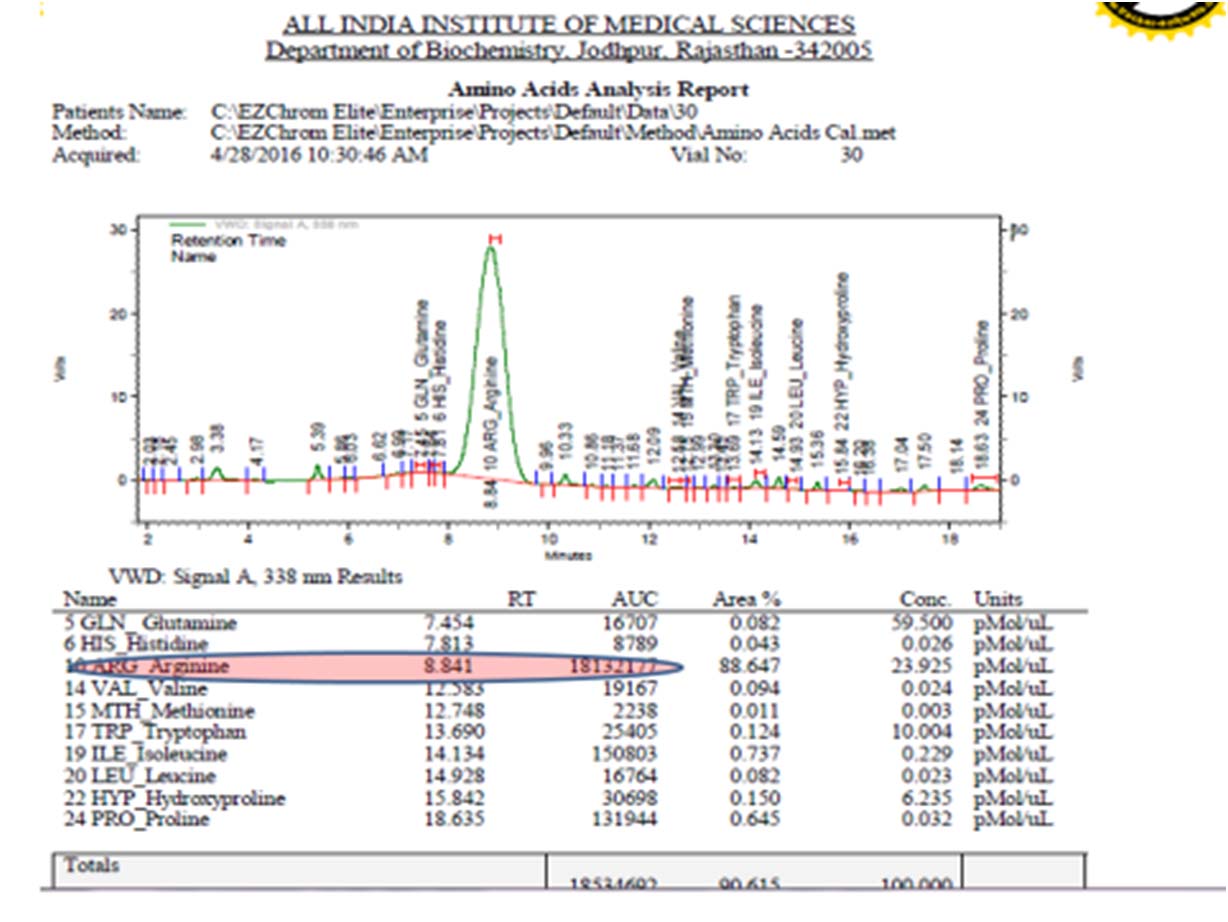
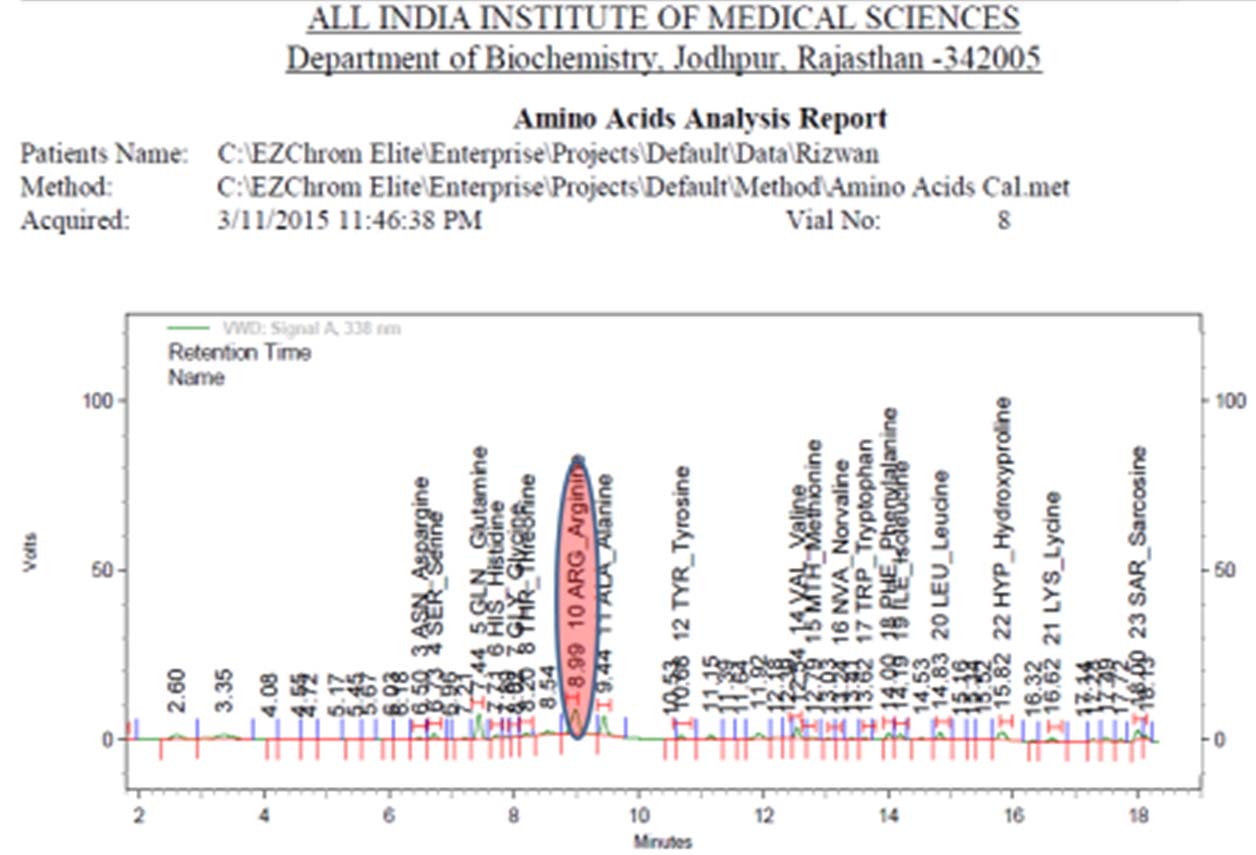
In addition to routine investigations, some special investigations to rule out inborn error of metabolism were also done. High sensitivity C Reactive Protein (Hs-CRP) was 8.89 mg/dl (normal range <1 mg/dl), plasma ammonia levels was 117.64 μmol/l (normal range 10-45 μmol/l) and plasma lactate level was 13 mg/dL (normal range 4.5-20 mg/dL). Plasma amino acid analysis revealed high serum arginine 387 μmol/l (normal range 39.5-42.5 μmol/l) and high serum glutamine 466 μmol/l (normal 322-400 μmol/l). Urinary Orotic Acid (OA)/creatinine ratio was 3.2 (normal-0.5-3.3 mmol/mole of creatinine) [Table/Fig-1,2].
Amino Acid Assay (AAA) on 1220 Agilent HPLC showing increased arginine peak in plasma (Sample cup 30).
Urine analysis for amino acid assay on 1220 Agilent HPLC system.
MRI head showed symmetric patchy regions of white matter abnormalities of periventricular areas and bilateral symmetric abnormalities of posterior limbs of internal capsules suggestive of inborn error of metabolism.
Child was kept on Intravenous (IV) antibiotic initially in view of fever and positive CRP. SAM management was started accordingly, feed given initially F-75 then F-100 and rest multivitamin given accordingly. Initially she was kept on IV antibiotics (ceftriaxone), in view of persistent fever, vomiting and feed intolerance. After initial improvement, patient deteriorated after seven days and clinically aspiration pneumonia was suspected. She was kept on IV fluid and injection vancomycin (20 mg/kg IV) was started. Patient improved after 3-4 days, feeding was restarted. After five days, she again developed fever, irritability and refusal to feed. Blood culture was done and IV antibiotic upgraded to injection meropenem, (40 mg/kg IV every eight hours for five days), in view of aspiration pneumonia.
She was discharged after 20 days of stay and was gaining weight and accepting feed well at the time of discharge. Dietary recommendation included low protein diet and special dietary supplements like calolipid.
On follow up, she continued to have recurrent episodes of fever and hyperammonaemia and unfortunately succumbed to death after three months of initial presentation.
Discussion
UCDs are a group of inborn errors of hepatic metabolism that affect the transfer of waste nitrogen into urea. The urea cycle has two main functions: the detoxification of waste nitrogen into excretable urea and the de novo biosynthesis of arginine [1]. The cycle consists of six sequential enzymatic steps for conversion of nitrogen derived from proteins to urea. All the enzyme deficiency disorders of urea cycle have been identified. Most of these disorders like N-acetyl glutamate synthase deficiency, carbamoyl phosphate synthetase I deficiency, argininosuccinate synthetase deficiency, argininosuccinate lyase deficiency and arginase I deficiency are inherited as an autosomal recessive, but Ornithine Transcarbamylase (OTC) deficiency is inherited as X-linked [2]. Any of these deficiencies can result in hyperammonaemia and accumulation of different metabolites. Each specific disorder usually presents in the newborn period or in early infancy with hyperammonaemic encephalopathy and hyperglutaminaemia except hyperargininaemia [3]. Hyperargininaemia occurs due to the deficiency of the enzyme arginase I (ARG1) which is predominantly found in liver, RBCs and salivary gland. Arginase II, another type of arginase, is found in renal tissues. Arginase hydrolyses L-arginine to ornithine and urea. Unlike other UCDs, symptoms of hyperargininaemia typically do not occur until the age of four years [4].
The present case is of ARG1 deficiency, presenting in early infancy with hyperammonaemia. The literature describes only seven such cases and these cases demonstrate a wide variation in neonatal presentation of ARG1 deficiency. Argininaemia is a disorder in which the clinical picture is quite different from the other disorders of urea cycle. It was first reported in 1965 by Prasad AN [5]. The disease is caused by complete absence of the activity of the enzyme arginase [6]. Most common genetic mutation associated with argininaemia is that of ARG1 gene located on chromosome 6 at band q23 [2,3]. A small number of mutations have been heterogeneous. They are often picked as abnormal, because of delayed developmental milestones. In the neonatal period, patients may have convulsions and repeated cyclic vomiting, anorexia, irritability and failure to thrive. They can present as spastic diplegia or quadriplegia. Usually, the patients have spastic toe walking gait, if they are able to walk with scissoring of lower limb [7]. Convulsions are regularly observed with associated EEG abnormalities. Patients may develop microcephaly and cerebral atrophy. Psychomotor impairment may be severe. Liver dysfunction has been reported in some cases of ARG1 deficiency but severe liver disease in form of cirrhosis or hepatic failure is rare [8].
An interesting biochemical abnormality noted in ARG1 deficiency is orotic aciduria which was also present in the proband [8]. There are two mechanisms proposed for orotic aciduria in ARG1 deficiency. According to the first one, there is a functional decrease in OTC activity as a result of low ornithine because of ARG1 deficiency and as per the second mechanism, it is as a back up phenomenon due to accumulation of distal substrates affecting OTC activity [8]. As a result, accumulated carbamoyl phosphate is shunted towards the pyrimidine biosynthetic pathway and leads to orotic aciduria [4]. Urinary OA is also increased in other enzyme defects viz., OTC deficiency, citrullinaemia, argininiosuccinic aciduria, mitochondrial DNA defects, occasionally in Rett’s syndrome, Lesch Nyhan syndrome and other disorders of pyrimidine metabolism. Direct derivatives of arginine like alpha keto guanidinovaleric acid, N-acetyl arginine and argininic acid are also found in urine in hyperargininaemia as well as guanidinoacetic and guanidinobutyric acids which are compounds formed by transamination [9].
The metabolic blocks, at times may not be complete in this disorder; therefore, arginine levels could be normal. The current modality for the management of hyperammonaemia includes arginine bolus and/or infusion [10] which is needed in most of the other UCDs as arginine provides citrulline which is further condensed with carbamoyl phosphate to quench ammonia and reduce its toxicity. We suggest that in light of ARG1 deficient cases with hyperammonaemia, though rare, the specific diagnosis be made even more urgently and if possible, before intravenous arginine therapy is initiated, to avoid any added insult to the case diagnosed. There are only few cases published in literature which are presented in [Table/Fig-3] [1,2,11-15].
Eight reported cases of hyperarginininaemia presenting in infancy.
| Authors | Presentation | Age reported |
|---|
| Present case | Seizures, poor feeding, global developmental delay, later pneumonia | 7 months |
| Schiff M et al., [11] | Respiratory distress, seizures | 3 weeks |
| Ghai SJ et al., [1] | Poor feeding, lethargy, seizures | 6 weeks |
| Picker JD et al., [12] | Poor feeding, lethargy, seizures | 3 days |
| Braga AC et al., [2] | Jaundice, cirrhosis, vomiting, palpable liver and spleen | 2 months |
| Jorda A et al., [13] | Tremor, seizures, jaundice, cirrhosis, vomiting | 5 days |
| Scholl-Bürgi S et al., [14] | Infection, seizures, poor feeding, lethargy | 36 days |
| Segawa Y et al., [15] | Vomiting, seizures, hypertonia | 18 days |
Conclusion
Here, we highlight the fact that, though rare, ARG1 deficiency with hyperammonaemia can present at an unusually early age either in infancy or neonatal period, much more than previously considered. Therefore, the above presentation in early infancy with hyperammonaemia and normal plasma lactate on repeated investigation, Type V UCD should be considered in listing the differential diagnosis, as recurrent hyperammonaemia of any grade is a risk factor for significant brain injury and associated mortality.
[1]. Ghai SJ, Nagamani SCS, Blaser S, Siriwardena K, Feigenbaum A, Arginase I deficiency: Severe infantile presentation with hyperammonaemia: more common than reported?Mol Genet Metab 2011 104(1-2):107-11. [Google Scholar]
[2]. Braga AC, Vilarinho L, Ferreira E, Rocha H, Hyperargininaemia presenting as persistent neonatal jaundice and hepatic cirrhosisJ Peds Gastro Nutr 1997 24:218-21. [Google Scholar]
[3]. Scaglia F, Lee B, Clinical, biochemical and molecular spectrum of hyperargininaemia due to arginase I deficiencyAm J Med Genet 2006 142:113-20. [Google Scholar]
[4]. Brosnan ME, Brosnan JT, Orotic acid excretion and arginine metabolismJ Nutr 2007 137:1656-61. [Google Scholar]
[5]. Prasad AN, Breen JC, Ampola MG, Rosman NP, Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature reviewJ Child Neurol 1997 12:301-09. [Google Scholar]
[6]. Schlune A, Vom Dahl S, Häussinger D, Ensenauer R, Mayatepek E, Hyperargininaemia due to arginase I deficiency: the original patients and their natural history, and a review of the literatureAmino Acids 2015 47(9):1751-62. [Google Scholar]
[7]. Christmann D, Hirsch E, Mutschler V, Collard M, Marescaux C, Colombo JP, Late diagnosis of congenital argininaemia during administration of sodium valproateRevue Neurologique 1990 146(12):764-66. [Google Scholar]
[8]. Coman D, Yeplito Lee J, Boneh A, New indications and controversies in arginine therapyClin Nutr 2008 27(4):489-96. [Google Scholar]
[9]. Cowley DM, Bowling FG, McGill JJ, van Dongen J, Morris D, Adult-onset arginase deficiencyJ Inherit Metab Dis 1998 21(6):677-78. [Google Scholar]
[10]. Morris SM Jr, Arginine metabolism: boundaries of our knowledgeJ Nutr 2007 137(6 Suppl 2):1602S-9S. [Google Scholar]
[11]. Schiff M, Benoist JF, Cardoso ML, Elmaleh-Berges M, Forey P, Santiago J, Early onset hyperargininaemia: a severe disorder?J Inherit Metab Dis 2009 32(Suppl1):S175-78. [Google Scholar]
[12]. Picker JD, Puga AC, Levy HL, Marsden D, Shih VE, Degirolami U, Arginase deficiency with lethal neonatal expression: evidence for the glutamine hypothesis of cerebral edemaJ Pediatr 2003 142(3):349-51. [Google Scholar]
[13]. Jordá A, Rubio V, Portolés M, Vilas J, García-Piño J, A new case of arginase deficiency in a Spanish maleJ Inherit Metab Dis 1986 9(4):393-97. [Google Scholar]
[14]. Scholl-Bürgi S, Sigl SB, Häberle J, Haberlandt E, Rostásy K, Ertl C, Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiencyJ Inherit Metab Dis 2008 31(Suppl 2):S323-28. [Google Scholar]
[15]. Segawa Y, Matsufuji M, Itokazu N, Utsunomiya H, Watanbe Y, Yoshino M, A long-term survival case of arginase deficiency with severe multicystic white matter and compound mutationsBrain Dev 2011 33(1):45-48. [Google Scholar]