Amyloidosis is a group of disease that is characterized by the deposition of extracellular abnormal proteinaceous material (amyloid), in various organs. Amyloidosis involving the liver is common and the radiological findings are often nonspecific. We present the case of a 40-year-old female who presented with abdominal pain. Ultrasound abdomen was reported as massive hepatomegaly with diffuse liver parenchymal disease. Bone marrow aspiration showed normomegaloblastic erythroid hyperplasia and plasma cells were within normal limits (5%). Also, amorphous, eosinophilic fragmented to smudgy material within the interstitium of cell trails was seen. Bone marrow biopsy and liver biopsy also showed similar kind of homogenous eosinophilic material. Both liver biopsy and bone marrow biopsy were subjected to special stains which confirmed the presence of amyloid. The patient did not have clinical or laboratory findings suggestive of any other organ involvement. Thus, we conclude that clinical and imaging presentations of amyloidosis are often nonspecific, hence biopsy is always required to confirm the diagnosis. Amyloid deposits on bone marrow aspiration are a rare occurrence and are often missed. It is an unusual sighting with very few studies mentioning its occurrence.
Case Report
A 40-year-old female presented with abdominal pain, progressive weakness and loss of appetite. On general physical examination, there was presence of pallor and clubbing. Per abdomen examination revealed massive hepatomegaly. Clinically, the differential diagnoses were infiltrative disorders and hematological malignancy.
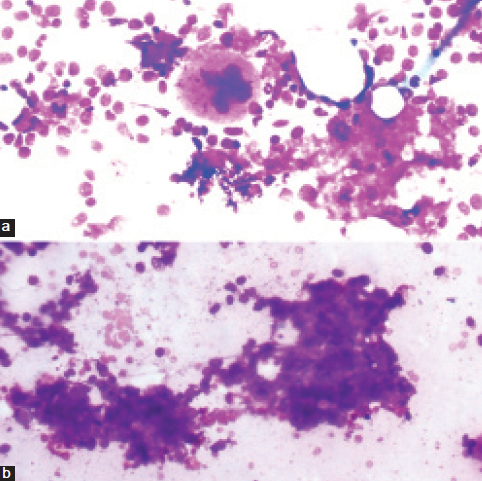
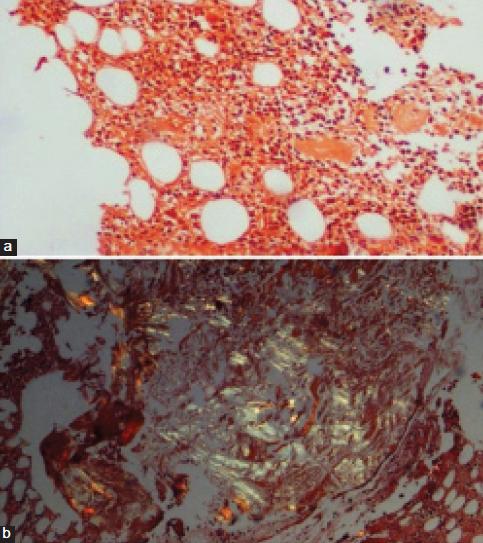
Ultrasound abdomen was reported as diffuse liver parenchymal disease with massive hepatomegaly with a span of 20 cm × 24 cm. Blood investigations revealed anaemia with reduced serum ferritin. Erythrocyte sedimentation rate was raised (80 mm/h). Serum calcium was within normal limits. Biochemical investigations revealed normal levels of serum total bilirubin. Serum direct bilirubin was slightly raised (0.69 mg/dl). Serum Alkaline Phosphatase (ALP) and serum Gamma-Glutamyl Transferase (GGT) were raised being 797 and 179 IU/L, respectively. Renal function tests were normal. Urine routine showed mild proteinuria (+1) in 24 hrs sample. Bence jones protein test was negative. Serum electrophoresis did not show M band. Subsequently, bone marrow aspiration was done with patient’s consent which showed hypercellular marrow with mild normomegaloblastic erythroid hyperplasia and plasma cells were within normal limits (5% on 500 differential count) [Table/Fig-1]. Also, there were few small aggregates of plasma cells (3-4 cells) and presence of amorphous, eosinophilic fragmented to smudgy material within the interstitium of cell trails was seen [Table/Fig-2]. Bone marrow biopsy showed hypercellular marrow with erythroid hyperplasia amidst which were seen patches of homogeneous eosinophilic material which was confirmed to be amyloid on Congo red staining and showed apple green birefringence on polarized microscopy [Table/Fig-3].
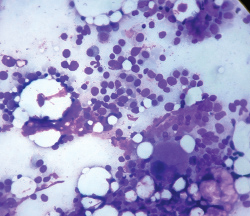
Bone marrow aspirate shows plasma cells along with other myeloid and erythroid precursors (Giemsa stain, 40X).
Bone marrow aspirate shows a) A single megakaryocyte along with amorphous, eosinophilic fragmented material; b) Acellular amorphous, eosinophilic fragmented to smudgy material within the interstitium of cell trail (Giemsa stain, 20X).
Bone marrow biopsy, a) Congophilic homogenous, amorphous material in the bone marrow (Congo red stain, 10X); b) Shows characteristic apple green birefringence of amyloid under the polarized microscope (Congo red stain, 10X).
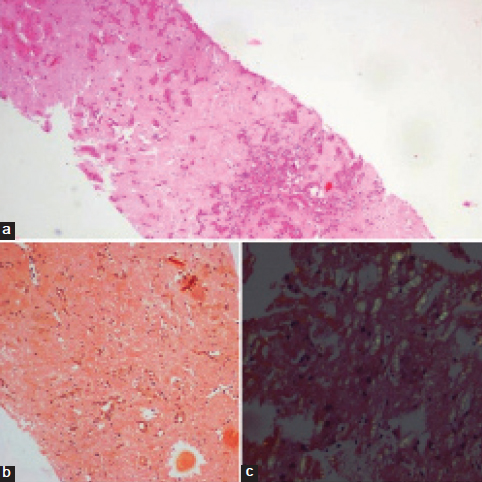
Subsequently, ultrasound guided liver biopsy was done which showed diffuse and extensive replacement of the liver parenchyma by homogenous eosinophilic material amidst which were seen hepatocytes, scattered singly and this was also confirmed as amyloid [Table/Fig-4]. There was a marked atrophy of liver parenchyma. Duodenal biopsy was also done that showed chronic nonspecific duodenitis.
Liver biopsy, a) Extensive replacement of liver parenchyma with homogenous, eosinophilic material with few preserved hepatocytes (H&E stain, 4X); b) Replacement of liver parenchyma with congo red positive homogenous material with few preserved hepatocytes (Congo red stain, 10X); c) Characteristic apple green birefringence of amyloid under the polarized microscope (Congo red stain, 40X).
A final diagnosis of primary amyloidosis was given. The patient was symptomatically treated for abdominal pain with analgesics and was eventually lost for follow up as she did not come back.
Discussion
Amyloidosis is a condition characterized by number of inherited and inflammatory disorders associated with extracellular deposits of fibrillar proteins that lead to tissue damage and functional compromise [1].
The two main forms of amyloidosis include primary amyloidosis (AL) and secondary amyloidosis (AA). Primary amyloidosis is associated with multiple myeloma and other B-cell dyscrasias. While, chronic disorders such as tuberculosis, rheumatoid arthritis, ankylosing spondylitis and long-standing malignancies, especially renal cell carcinoma lead to secondary amyloidosis [2].
Accumulation of large amount of amyloid leads to organomegaly. Grossly, the organ becomes grey with a firm and waxy consistency.
Histologically, amyloid deposition is always extracellular and usually begins in between the cells. Consequently, surrounding the cells and destroying them [1].
Our patient presented with vague symptoms of abdominal pain, weakness and loss of appetite. Per abdomen examination revealed massive hepatomegaly. Clinically, a provisional diagnosis of infiltrative liver disease was given, the cause of which was uncertain.
Among the battery of tests done in our case, the first clue to the presence of amyloid was clinched by the bone marrow aspirate which showed the presence of homogenous, smudgy, eosinophilic deposits. This is a rare occurrence. Such an occurrence has not been mentioned in most of the textbooks; hence, most of these cases are missed on bone marrow aspiration [3].
One such case has been reported by Ahuja A et al., [4], where the bone marrow aspirate in a suspected case of amyloidosis showed acellular, pink deposits of amyloid. The plasma cells were approximately 6%. However, in our case, the plasma cell count was 5%. There were few isolated clusters of plasma cells (3-4 cells per cluster). In a study done by Swan N et al., [5], it is stated that in AL amyloidosis, the distribution of neoplastic plasma cells is subtle and can be seen singly or in isolated small clusters which were similar to that seen in our case. In their study, the majority of the cases had low plasma cell count (<10% of the total marrow cellularity).
Furthermore, the presence of amyloid in our study was confirmed on bone marrow biopsy which showed eosinophilic, homogenous, acellular deposits of amyloid which was confirmed by Congo red staining and apple green birefringence on polarized microscopy.
Subsequently, a biopsy done from the liver showed similar deposits of amyloid. The liver is commonly involved in secondary as well as primary amyloidosis and presents as massive hepatomegaly [6]. Extensive amyloid deposition in the liver in patients with systemic amyloidosis has also been demonstrated in a similar study done by Chopra S et al., [7], where histopathological examination of the liver biopsy revealed amyloid deposition in all 38 patients with systemic amyloidosis.
In our case, the patient presented with increased levels of ALP and gamma glutamyltransferase suggestive of cholestasis, secondary to amyloidosis which is characteristic of such condition [8].
Subsequently, a biopsy was done from the duodenum to rule out the involvement of other gastrointestinal organs by amyloidosis or secondary causes of amyloidosis such as ulcerative colitis and Crohns disease. The biopsy showed chronic nonspecific duodenitis.
In our case, neither the urine showed the presence of bence jones proteins nor the serum electrophoresis showed the presence of M band. The calcium levels were within the normal limits. Moreover, the plasma cell count in the bone marrow aspirate was not significantly raised (5%). The history, clinical examination and other haematological, biochemical and histopathological investigations did not suggest the presence of any chronic inflammatory condition or malignancy.
Based on these findings, a clinicopathological diagnosis of systemic amyloidosis was given. It was not ascertained whether amyloidosis was primary or secondary.
Primary amyloidosis can present with a varied plasma count which can be as high as seen in the cases of overt multiple myeloma (>60%) or the count can be <10% in which case it is classified as monoclonal gammopathy of unknown significance. In such cases, bone marrow aspirate usually does not show amyloid; however, biopsy shows amyloid deposition in the vessel wall and in the interstitium [4].
Moreover, the total plasma cell count in the bone marrow aspirate can be <10% in cases of AL amyloidosis [5].
Amyloidosis was considered to be primary only in the absence of associated disease [7].
In our case, in the absence of a history of chronic disorders, the presence of mild proteinuria and presence of isolated small clusters of plasma cells in the bone marrow, presenting with extensive amyloid deposits in the liver and bone marrow, primary amyloidosis can be suspected probably due to occult plasma cell dyscrasia. However, immunohistochemical stains for light chains immunoglobulins could have helped to establish monoclonal gammopathy which was not possible in our case as the patient could not afford it.
Conclusion
Hence, we conclude that primary amyloidosis is a rare entity and even rarely presents with normal plasma cell count. Very few cases have been reported in the literature with bone marrow aspirate smears showing amyloid deposits. Our case is one of those rare cases. In such cases, extensive histopathological examination of the bone marrow along with other organs such as liver and spleen using special stains is required to reach a diagnosis of primary amyloidosis.
[1]. Kumar V, Abbas AK, Aster JC, Diseases of the immune systemRobbins and Cotran Pathologic Basis of Disease 2014 9th edPhiladelphia, PAWB. Saunders Co:256-57. [Google Scholar]
[2]. Srinivasan S, Tan YQ, Teh HS, Lee PJ, Khoo RN, Primary hepatic amyloidosis presenting as nodular masses on the background of diffuse infiltration and extreme liver stiffness on MR elastographyJ Gastrointestin Liver Dis 2014 23(4):437-40. [Google Scholar]
[3]. Chopra A, Anand M, Kumar R, Kalita D, Raina V, Unusual sighting: Amyloid deposits in bone marrow aspirate in multiple myelomaIndian J Pathol Microbiol 2008 51(4):562-63. [Google Scholar]
[4]. Ahuja A, Prabhat P, Rastogi A, Panda D, Kumar CK, Sarin SK, Extensive amyloid deposit in bone marrow aspirateIndian J Hematol Blood Transfus 2013 29(3):187-88. [Google Scholar]
[5]. Swan N, Skinner M, O’Hara CJ, Bone marrow core biopsy specimens in AL (primary) amyloidosis. A morphologic and immunohistochemical study of 100 casesAm J Clin Pathol 2003 120(4):610-16. [Google Scholar]
[6]. Jeong HJ, Hahn EK, Kim E, Park CI, Hepatic amyloidosis - Two cases reportJ Korean Med Sci 1988 3(4):151-55. [Google Scholar]
[7]. Chopra S, Rubinow A, Koff RS, Cohen AS, Hepatic amyloidosis. A histopathologic analysis of primary (AL) and secondary (AA) formsAm J Pathol 1984 115(2):186-93. [Google Scholar]
[8]. Cichoz-Lach H, Prozorow-Król B, Swatek J, Skrzydlo-Radomanska B, Buk L, Zdunek M, Hepatomegaly, weight loss and general malaise - The first manifestations of primary systemic amyloidosisPrz Gastroenterol 2014 9(1):57-61. [Google Scholar]