Crigler Najjar Syndrome Type 2 (CNS Type 2): An Unwonted Cause of Jaundice in Adults
Prabhat Kumar1, Gargi Sasmal2, Shreya Gupta3, Renu Saxena4, Sudha Kohli5
1 Senior Resident, Department of General Medicine, Dr. Ram Manohar Lohia Hospital, New Delhi, India.
2 Postgraduate Student, Department of General Medicine, Dr. Ram Manohar Lohia Hospital, New Delhi, India.
3 Postgraduate Student, Department of General Medicine, Dr. Ram Manohar Lohia Hospital, New Delhi, India.
4 Consultant, Department of Genetic Medicine, Sir Ganga Ram Hospital, New Delhi, India.
5 Consultant, Department of Genetic Medicine, Sir Ganga Ram Hospital, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Gargi Sasmal, Postgraduate Student, Department of General Medicine, Dr. Ram Manohar Lohia Hospital, Baba Kharak Singh Marg, New Delhi, India.
E-mail: gargisplasharoma@yahoo.co.in
Crigler Najjar Syndrome (CNS) Type 2 is an uncommon genetic disorder characterised by non-haemolytic unconjugated hyperbilirubinemia. It is caused by mutations in the UGT1A1 gene which codes for the enzyme uridine diphosphate glucoronosyl transferase- 1, required for the conjugation and further excretion of bilirubin from the body. Affected individuals are usually asymptomatic apart from the jaundice and investigations reveal isolated indirect hyperbilirubinemia. It can be conveniently diagnosed by evaluating the response to phenobarbitone in terms of fall in bilirubin levels. Genetic testing of the UGT1A1 gene for mutations is the diagnostic clincher. However, case reports documenting the genetic mutational analysis are sparse. We report one such rare case.
Gilbert’s syndrome, Isolated indirect hyperbilirubinemia, Phenobarbitone
Case Report
A 28-year-old female with history of normal vaginal delivery one month back presented with complaints of yellowish discolouration of eyes and body since childhood which had exacerbated for the last two months. During the eighth month of her latest pregnancy, she had developed fever which was of moderate grade but subsided within three days after taking some antipyretics. This episode was however associated with an increase in the already present jaundice. The increased jaundice remained as such and showed no signs of a declining trend. There was no history of weight loss, malaise, alteration in stool colour or consistency, itching over body, vomiting, bleeding from any site or abdominal pain. Neither, was there any history of neuropsychiatric complaints, blood transfusion, chronic drug exposure or any addictions. She was born of a non consanguineous marriage and there were no similar complaints in any of the family members. On physical examination, she had pallor and icterus with no signs of liver cell failure. The systemic examination was grossly normal.
Investigations showed hemoglobin of 7.5 g/dl, total leukocyte count of 6,300/mm3 and a platelet count of 3.4 lac/mm3. Mean Corpuscular Volume (MCV) was 67 fL with a corrected reticulocyte count of 3.2%. The peripheral smear was suggestive of microcytic hypochromic anemia. Liver function tests showed a total bilirubin of 20 mg/dl, of which the indirect component was 16.7 mg/dl and direct fraction was 3.3 mg/dl. Liver enzymes were essentially normal and there was no evidence of any coagulopathy. Other biochemical parameters including renal function tests, serum electrolytes and serum protein were within normal limits. Blood tests for hepatitis A, B, C, E and HIV were negative. There was no evidence of any ongoing hemolysis as confirmed by normal serum Lactate Dehydrogenase (LDH) and haptoglobin levels. The iron profile was indicative of iron deficiency anemia with a low serum ferritin and iron levels along with an elevated Total Iron Binding Capacity (TIBC). Stool evaluation was negative for occult blood and parasites or cysts. Ultrasonography of the abdomen and Upper Gastrointestinal Endoscopy (UGIE) were also normal.
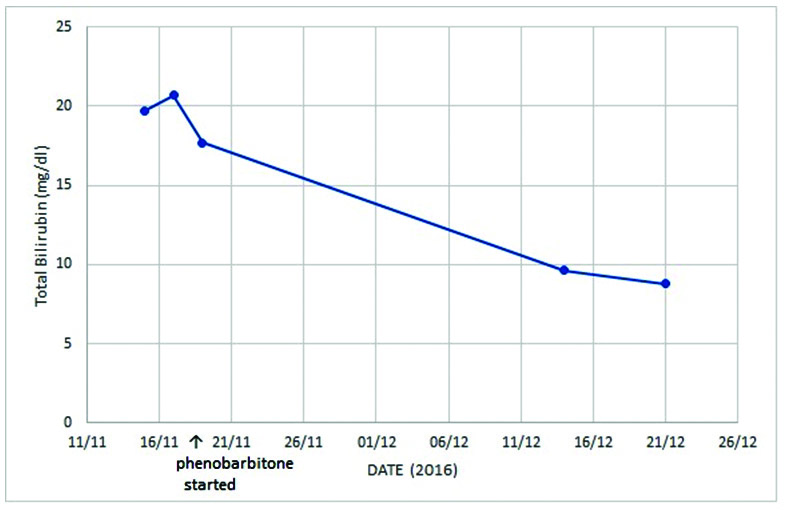
In view of the significant longstanding jaundice since childhood and isolated indirect hyperbilirubinemia, patient was suspected to have a genetic disorder of bilirubin conjugation. The high bilirubin levels went more in favour of CNS. Hence, the patient was started empirically on oral phenobarbitone at a dose of 60 mg thrice daily. Meanwhile, her blood sample was sent for genetic mutational analysis. Patient was then followed up four weeks later. Post phenobarbitone therapy her total bilirubin levels fell sharply by more than 50% to 9.62 mg/dl with an indirect fraction of 8.32 mg/dl [Table/Fig-1]. This exorbitant fall in bilirubin levels was suggestive of CNS Type 2. Further, the genetic sequencing and analysis of the UGT1A1 gene showed that the patient was compound heterozygous for p.G276R and -85 to -83 CAT insertion in promoter region, which confirmed the diagnosis. The patient was advised genetic counseling.
Significant fall in bilirubin levels following initiation of treatment with phenobarbitone.
Discussion
Crigler Najjar Syndrome (CNS) includes two categories; type 1 and type 2. In both the types, mutations in the UGT1A1 gene is the underlying defect. Type 1 CNS, the perilous variety, caused by the complete lack of the enzyme, presents since birth with newborns rarely surviving beyond infancy, succumbing to bilirubin encephalopathy. Type 2 CNS also known as Arias syndrome, named after the physician who first described it in 1962, comes with only a partial absence of the enzyme and is a less severe form with affected patients having a far better life expectancy [1]. Crigler Najjar syndrome is majorly autosomal recessive although variations are known to occur in the inheritance pattern of type 2 CNS [2]. The prevalence of CNS type 2 is not known but an approximate annual incidence of 1 per million live births has been found in case reports from worldwide for both the types of CNS [3].
CNS type 2 presents with persistent jaundice that is exacerbated by intercurrent illness, stress, pregnancy or drug use. Jaundice may also occur in infancy or later in childhood. Examination is essentially normal, apart from icterus. This is further confirmed by normal imaging studies. Serum bilirubin levels in these cases is usually upto 20 mg/dl, with increments to approximately 40 mg/dl during exacerbations [4]. Hence, it is differentiated facilely from its doppelgangers-Haemolytic syndromes and Gilbert’s syndrome in which bilirubin levels rarely cross 6 mg/dl [5]. No other biochemical parameter apart from serum bilirubin is deranged. In case of a debatable diagnosis where the bilirubin levels are only marginally raised, along with slight increases in serum LDH levels and normal serum haptoglobin levels, the genetic disorders of bilirubin conjugation first need to be precisely differentiated from other causes that can cause haemolytic jaundice in adulthood [6]. Such patients have to undergo certain specific tests to rule out congenital hemoglobinopathies (like thalassemia and sickle cell disease), autoimmune haemolytic anaemias (AIHA), RBC membrane defects, Thrombotic Microangiopathy (TMA) and Paroxysmal Nocturnal Hemoglobinuria (PNH). These specific tests include haemoglobin electrophoresis, coomb’s test (direct and indirect), osmotic fragility test, a detailed peripheral smear to look for specific markers of hemolysis like schistocytes and spherocytes and CD55 and CD59 assays. In our case, the very high bilirubin level, in the first place ruled out haemolytic disorders. Therefore, all these specific tests were not done in our patient. The next hurdle is to differentiate the three syndromes: Gilbert’s, CNS type 1 and CNS type 2. The most reasonable available approach is to evaluate the fall of bilirubin levels in response to phenobarbitone administration. In Gilbert’s, following phenobarbitone administration, the bilirubin levels completely normalise. In CNS type 2 the fall in bilirubin levels is usually more than 30 percent, but the levels never normalise. In CNS type 1, the fall is almost nil. The response in CNS type 2 with phenobarbitone is due to induction of the already present residual 10%-30% of (UGTA) enzyme activity required for bilirubin conjugation [7]. Hence, theoretically and therapeutically, as an alternative, drugs like phenytoin, phenazone can also be used [8]. In the still remaining few patients with a questionable diagnosis, analysis of bile pigments for bilirubin conjugates by chromatography can clinch the diagnosis. A significant quantity of conjugated bilirubin (mostly monoconjugates) is observed in CNS type 2, whereas it is absent or detected only in trace amounts in patients of CNS type 1. Molecular and genetic testing is available in very few centres. In India, only two cases of CNS type 2 with mutational analysis have been reported, both of which were homozygous for the genetic polymorphisms attributing to the disease. In our patient, the mutation analysis showed that the patient was compound heterozygous for the affected genetic loci. This is probably the first case report of its kind.
Treatment of CNS type 2 is usually conservative with avoidance of drugs that displace bilirubin from albumin like penicillin, sulphonamides, salicylates, ceftriaxone and furosemide [9]. Also, lifelong administration of phenobarbitone is advocated to prevent kernicterus even though the risk is minimal [9]. The dose of phenobarbitone is 3-5 mg/kg/day titrated preferably to 60- 180 mg/ day in single or divided doses [10]. The dose is reduced further (30- 60 mg/ day) in pregnancy to avoid its teratogenic side effects. The response is seen within two to three weeks. A favourable substitute with similar efficacy and fewer adverse effects is clofibrate (2 g/day in divided doses), but it is contraindicated in pregnancy [11]. Adjuvant calcium supplementation has also been found to increase the gut excretion of bilirubin [12]. The role of plasmapheresis or phototherapy is reserved to tide over a hyperbilirubinemic crisis, especially in pregnancy so as to prevent the transplacental transit of bilirubin and subsequent neurological damage to the fetus. Rarely, patients require exchange transfusions or long term phototherapy. Newer therapeutic modalities such as hepatocyte transplantation and gene therapy have not been used for CNS type 2 till date as the affected patients thrive well on oral phenobaritone only. Genetic counseling should also be done for the affected individuals and their families.
Conclusion
CNS Type 2 is rare genetic disorder of bilirubin metabolism and all the more, a rarer cause of jaundice in an adult. Yet, a high level of clinical suspicion based on some of its unique features can avoid many extraneous and costly investigations. Hence, a patient presenting with asymptomatic indirect hyperbilirubinemia should be evaluated for CNS.
[1]. Radlovic N, Hereditary hyperbilirubinemiasSrpski arhiv za celokupno lekarstvo 2014 142(3-4):257-60. [Google Scholar]
[2]. Chaudhary R, Maheshwari A, Deepmala D, Tapparwal V, A rare case of Crigler-Najjar syndrome type II with pregnancyInt J Reprod Contracept Obstet Gynecol 2014 3(1):261-62. [Google Scholar]
[3]. Maruo Y, Nakahara S, Yanagi T, Nomura A, Mimura Y, Matsui K, Genotype of UGT1A1and phenotype correlation between Crigler-Najjar syndrome type II and Gilbert syndromeJournal of Gastroenterology and Hepatology 2016 31(2):403-08. [Google Scholar]
[4]. Mori Y, Kimura H, Suehiro K, Naritomi Y, Maeda Y, Kusaba T, A case of II type Crigler-Najjar syndromeNihon Naika Gakkai Zasshi 1991 80(1):102-03. [Google Scholar]
[5]. Blueger AF, Krupnikova EZ, Sondore VY, Semushina EP, Study of the etiology and pathogenesis of low grade nonhemolytic unconjugated hyperbilirubinemia (Gilbert’s disease)Acta Hepatogastroenterol (Stuttg) 1977 24:140 [Google Scholar]
[6]. Pratt D, Kaplan M, Evaluation of abnormal liver-enzyme results in asymptomatic patientsNew England Journal of Medicine 2000 342(17):1266-71. [Google Scholar]
[7]. Van der Veere CN, Sinaasappel M, McDonagh AF, Rosenthal P, Labrune P, Odiévre M, Current therapy for Crigler-Najjar syndrome type 1: Report of a world registryHepatology 1996 24(2):311-15. [Google Scholar]
[8]. Hafkamp A, Nelisse-Haak R, Sinaasappel M, Oude Elferink RP, Verkade HJ, Orlistat Treatment of unconjugated hyperbilirubinemia in Crigler-Najjar disease: a randomized controlled trialPediatric Research 2007 62(6):725-30. [Google Scholar]
[9]. Nair KM, Lohse P, Nampoothiri S, Crigler-Najjar syndrome type 2: Novel UGT1A1 mutationIndian Journal of Human Genetics 2012 18(2):233-34. [Google Scholar]
[10]. Ranjan P, Kohli S, Saxena R, Thakur S, Mutation analysis in Crigler-Najjar Syndrome type ii—case report and literature reviewJournal of Clinical and Experimental Hepatology 2011 1(3):204-06. [Google Scholar]
[11]. Passuello V, Puhl AG, Wirth S, Steiner E, Skala C, Koelbl H, Pregnancy outcome in maternal Crigler-Najjar syndrome type II: a case report and systematic review of the literatureFetal Diagn Ther 2009 26:121-26. [Google Scholar]
[12]. Pett S, Mowat AP, Crigler-Najjar syndrome types I and II. Clinical experience—King’s College Hospital 1972-1978. Phenobarbitone, phototherapy and liver transplantationMol Aspects Med 1987 9(5):473-82. [Google Scholar]