Splenic Marginal Zone Lymphoma (SMZL) is a rare B-cell neoplasm comprising less than 2% of non-Hodgkin lymphomas. We hereby report a case of SMZL in a 66-year-old female who presented with fever and massive splenomegaly. Peripheral blood smear examination showed atypical lymphoid cells showing variable cytoplasmic processes. Flowcytometric immunophenotyping of peripheral blood showed tumour cells which were found to be positive for CD19, CD79b and showing kappa light chain restriction along with lack of expression for CD5, CD10, CD23, CD103 and lambda. These findings were suggestive of B cell chronic lymphoproliferative disorder. Various differential diagnoses considered in this case were analysed by using different diagnostic clues to arrive at the diagnosis.
Bone marrow examination and Immunohistochemical (IHC) analysis showed tumour cells in nodular, interstitial and intrasinusoidal pattern of infiltration which were positive for CD20 and CD79b with kappa light chain restriction and lack of expression of CD5, CD10, CD23 and CD103 which further corroborated the flowcytometric immunophenotyping. The diagnosis of SMZL is arrived at by a combination of diagnostic clues like clinical features, peripheral smear findings, flowcytometric immunophenotyping, morphological and IHC findings in bone marrow biopsy. This case highlights the significance of flowcytometric immunophenotyping and bone marrow biopsy with immunohistochemistry to arrive at a diagnosis of SMZL even in absence of splenic histopathology.
CD20, Intrasinusoidal pattern, Massive splenomegaly
Case Report
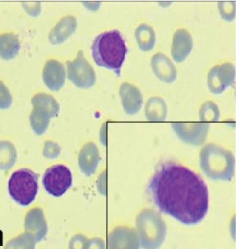
A 66-year-old female presented with easy fatiguibility, generalised weakness, exertional dyspnoea, fever on and off since five to six months and left hypochondrial pain since 15 days. On examination, massive splenomegaly with mild hepatomegaly was noted, however no lymphadenopathy was found. Ultrasound abdomen showed gross splenomegaly and mild hepatic enlargement. Biochemical investigations revealed deranged renal function test: raised urea 102 mg/dl and creatinine 1.7 mg/dl. Liver function test was found to be within normal limits. Complete blood count showed haemoglobin 5.7 gm/dl, White Blood Cells (WBC) count 15,200/cumm (Polymorphs-16%, Lymphocytes-82%, Eosinophils-2%), platelet count 1.5 lac/µl and ESR 28 mm/hr. Peripheral blood smear showed 82% lymphoid cells which were small to intermediate in size, having condensed chromatin, few with evident nucleoli and scanty to moderate amount of pale blue cytoplasm. Many of the cells showed variable number of cytoplasmic processes [Table/Fig-1].
Peripheral blood smear showing atypical lymphoid cells with cytoplasmic processes {villous lymphocytes} (Giemsa, 40X); Inset: atypical lymphoid cells displaying polar villous processes (Giemsa, 100X).
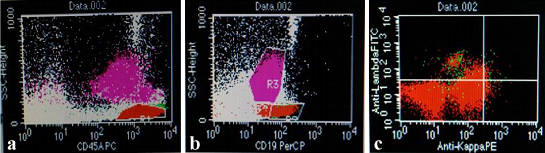
Flowcytometric immunophenotyping of peripheral blood revealed tumour cells to be positive for CD19 and CD 79b and showing kappa light chain restriction [Table/Fig-2a-c]. Tumour cells were found to be negative for CD5, CD10, CD23, CD103 and lambda.
a) CD45/SSC scattergram revealed an abnormal lymphoid cell cluster showing moderately bright CD45 positivity; b) CD19/SSC scattergram showing CD19 positivity; c) Lymphoid cells showing Kappa restriction.
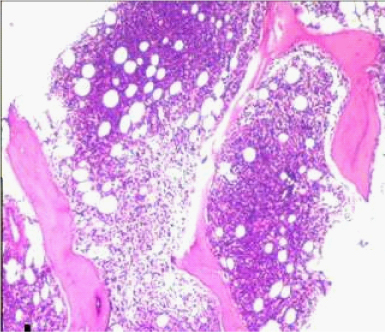
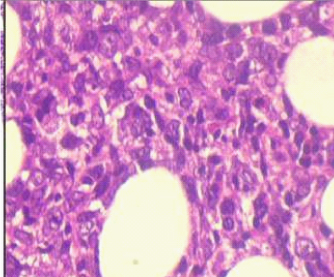
Bone marrow aspiration smears showed haemodilution, however imprint smears showed marked prominence of lymphoid cells constituting 70% of all nucleated cells. Bone marrow biopsy showed almost complete replacement by abnormal lymphoid cells arranged in vague nodular pattern, interstitial and intrasinusoidal pattern. It comprised of mainly small lymphoid cells having condensed chromatin and scant cytoplasm. The other population was of intermediate sized cells showing nuclear indentation and scant cytoplasm (possibly centrocyte-like cells). A significant number of intermediate sized cells showed moderate amount of pale eosinophilic cytoplasm (monocytoid cells). Interspersed in between were scattered large cells with evident nucleoli and cells showing plasmacytic differentiation [Table/Fig-3,4].
Photomicrograph of bone marrow biopsy showing nodular and interstitial pattern of infiltration (H&E 10X).
Atypical lymphoid cells of varied morphology (centrocyte like and monocytoid cells) (H&E 40X).
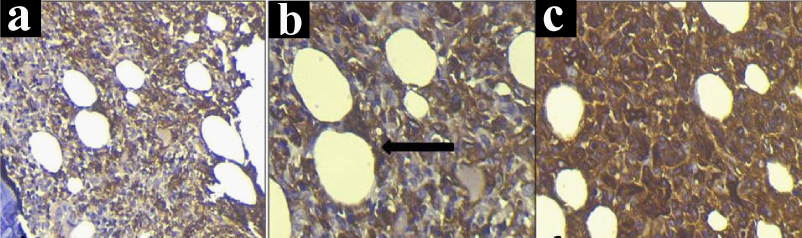
On immunohistochemistry, pattern of infiltration was better highlighted by CD20 immunostaining. It showed nodular, interstitial and intrasinusoidal pattern of infiltration. Immunohistochemistry corroborated the flowcytometric findings of tumour cells showing kappa light chain restriction [Table/Fig-5a-c]. These cells were similarly found to be negative for CD5, CD10, CD23 and lambda. Peripheral blood, bone marrow aspiration and bone marrow biopsy findings in conjunction with immunohistochemistry and flowcytometric immunophenotyping were found to be consistent with the diagnosis of SMZL. In view of poor general condition, the treatment could not be started, however the patient’s condition got worsened which led to her demise.
a) Immunostaining showing CD20 positive cells in nodular and interstitial pattern of infiltration (IHC, 20X); b) Immunostaining showing CD20 positive cells, arrow showing intrasinusoidal pattern of infiltration (IHC, 40X); c) Immunostaining showing kappa light chain restriction (IHC, 40X).
Discussion
SMZL is an uncommon low grade B-cell neoplasm accounting for less than 2% of all non-Hodgkin lymphomas and only 8.3% of all non-Hodgkin’s lymphoproliferative disorders involving spleen [1]. It is a rare indolent B-cell neoplasm involving the spleen, liver, bone marrow and blood [2]. It usually presents in elderly age [3]. Clinically, the patient presents with fever, generalised weakness, left hypochondrial pain due to moderate to massive splenomegaly. Hepatomegaly is relatively less common; however, lymphadenopathy is extremely rare. B-symptoms such as fever and night sweats are rare [3]. The diagnosis requires splenic histology or peripheral smear examination; however, with advent of immunophenotyping, splenectomy is no longer mandatory for diagnosis [4].
The diagnosis of SMZL requires careful interpretation of all the diagnostic clues. The differential diagnoses considered in this case were SMZL, Hairy Cell Leukaemia (HCL), Hairy Cell Leukaemia – Variant (HCL-V), Splenic Diffuse Red Pulp Lymphoma (SDRPL), small B cell non-Hodgkin’s lymphoma like Small Cell Lymphoma (SLL)/Chronic Lymphocytic Leukaemia (CLL), follicular lymphoma, mantle cell lymphoma and lymphoplasmacytic lymphoma. The immunophenotypic findings of SMZL and other lymphoproliferative disorders have been shown in [Table/Fig-6] [4].
Immunophenotypic analysis of SMZL and other B-cell lymphoproliferative disorders.
| CD5 | CD19 | CD20 | CD23 | CD79b | CD10 | CD103 |
|---|
| Our case (SMZL) | - | + | + | - | + | - | - |
| HCL | - | + | + | - | + | - | + |
| HCL-v | - | + | + | - | + | - | -/+ |
| SDRPL | -/+ | + | + | - | + | - | - |
| CLL/SLL | + | + | -/+ | + | + | - | - |
| FL | - | + | + | -/+ | + | + | - |
| MCL | + | + | -/+ | -/+ | + | - | - |
| LPL | - | + | + | - | + | - | - |
SMZL/SLVL: Splenic Marginal Zone Lymphoma/Splenic Lymphoma With Villous Lymphocytes; +: positive; -: negative; +/-: weakly positive; HCL: Hairy Cell Leukaemia; HCL-v: Hairy Cell Leukaemia Variant, SDRPL: Splenic Diffuse Red Pulp Lymphoma; CLL/SLL: Chronic Lymphocytic Leukaemia/Small Lymphocytic Lymphoma; MCL: Mantle Cell Lymphoma; FL: Follicular Lymphoma; LPL: Lymphoplasmacytic Lymphoma
In classic hairy cell leukaemia, recurrent opportunistic infections, atypical lymphoid cells with circumferential villous projections, mature nuclear chromatin without prominent nucleoli are seen. Peripheral blood shows pancytopenia. Bone marrow biopsy shows diffuse reticulin fibrosis with presence of cells having abundant cytoplasm and perinuclear clearing giving a “Fried- egg” appearance. Immunophenotypic profile shows B cell lymphoid neoplasm with CD 103 expression. Absence of these features excludes this possibility [5].
HCL-V is usually associated with characteristic prominent nucleoli in the tumour cells. Lack of this cytomorphological feature excludes this possibility. In SDRPL, diffuse infiltration of splenic pulp and bone marrow is seen, however, absence of erythematous and pruritic cutaneous papules with paucity of lymphoid follicles in bone marrow biopsy make this extremely rare diagnosis unlikely [5].
Among small B cell lymphoid neoplasm, lack of lymphadenopathy, presence of villous processes and lack of expression of CD5 and CD23 in lymphoid cells excludes a diagnosis of B-CLL. Follicular lymphoma was excluded by lack of CD10 expression and absence of characteristic paratrabecular pattern of infiltration in the bone marrow. Mantle cell lymphoma was also excluded by the absence of lymphadenopathy and lack of CD5 expression in the tumour cells. Differentiation of Lymphoplasmacytic Lymphoma (LPL) from SMZL was quite challenging specially on bone marrow biopsy. Both SMZL and LPL display plasmacytic differentiation, however, absence of lymphadenopathy and presence of monocytoid B cells excluded the diagnosis of LPL. Clinically, SMZL is associated with moderate to massive splenomegaly, however presence of peripheral lymphadenopathy is extremely uncommon [6].
Peripheral blood findings of anaemia and thrombocytopenia are due to hypersplenism. In addition, peripheral blood shows lymphocytosis comprising of small B lymphoid cells with villous processes. Bone marrow biopsy in our case revealed CD20 positive lymphoma cells arranged in nodular, interstitial and intrasinusoidal pattern of infiltration. SMZL infiltration into bone marrow has been reported in 67-100% of the cases at the time of diagnosis [7].
SMZL is not associated with any specific immunophenotypic profile and hence its diagnosis needs to be directed to exclude other subtypes. Correlation with other diagnostic clues is pertinent to arrive at a diagnosis. Molecular dysregulation of NF-kB, NOTCH2 and KLF2 genes is associated with SMZL lymphomagenesis. These mutated genes are associated with 35%, 40% and 20-40% of cases of SMZL respectively [1].
Asymptomatic patients do not require any treatment. Therapy is indicated only in symptomatic patients. Nowadays, splenectomy is usually not done and rituximab monotherapy is used as a firstline therapy in treatment of SMZL. In patients with disseminated disease, rituximab along with chemotherapy (cyclophosphamide, vincristine, doxorubicin and prednisone) is indicated [2].
Conclusion
SMZL is a rare B-cell lymphoid neoplasm involving spleen, bone marrow and frequently blood. Its diagnosis requires utmost attention of important clues especially in the absence of splenic histopathology for the evaluation. Presence of classical cell morphology (polar villous processes), flowcytometric immunophenotyping and typical nodular, interstitial and intrasinusoidal pattern of infiltration by CD20 positive cells in the bone marrow are important diagnostic clues for the diagnosis of SMZL. This case also highlights the importance of flowcytometric immunophenotyping and bone marrow biopsy with immunohistochemistry to exclude other possibilities and arrive at a definitive diagnosis. It is pivotal in the management of the patient as this will guide the haemato-oncologist for a precise therapeutic intervention.
SMZL/SLVL: Splenic Marginal Zone Lymphoma/Splenic Lymphoma With Villous Lymphocytes; +: positive; -: negative; +/-: weakly positive; HCL: Hairy Cell Leukaemia; HCL-v: Hairy Cell Leukaemia Variant, SDRPL: Splenic Diffuse Red Pulp Lymphoma; CLL/SLL: Chronic Lymphocytic Leukaemia/Small Lymphocytic Lymphoma; MCL: Mantle Cell Lymphoma; FL: Follicular Lymphoma; LPL: Lymphoplasmacytic Lymphoma
[1]. Brox A, Bishinsky JI, Berry G, Primary non-Hodgkin lymphoma of the spleenAm J Hematol 1991 38(2):95-100. [Google Scholar]
[2]. Arcaini L, Rossi D, Paulli M, Splenic marginal zone lymphoma: from genetics to managementBlood 2016 127(17):2072-81. [Google Scholar]
[3]. Franco V, Florena AM, Iannitto E, Splenic marginal zone lymphomaBlood 2003 101(7):2464-72. [Google Scholar]
[4]. Behdad A, Bailey NG, Diagnosis of splenic B-cell lymphomas in the bone marrow: a review of histopathologic, immunophenotypic, and genetic findingsArch Pathol Lab Med 2014 138(10):1295-301. [Google Scholar]
[5]. Swerdlow SH, Campo E, Harris LN, Jaffe ES, Pileri SA, Stein H, International Agency for Research on Cancer, World Health OrganizationWHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 2008 4th edLyon, FranceInternational Agency for Research on Cancer [Google Scholar]
[6]. Parry-Jones N, Matutes E, Gruszka-Westwood AM, Swansbury GJ, Wotherspoon AC, Catovsky D, Prognostic features of splenic lymphoma with villous lymphocytes: a report on 129 patientsBr J Haematol 2003 120:759-64. [Google Scholar]
[7]. Dierlamm J, Wlodarska I, Michaux L, Stefanova M, Hinz K, Van Den Berghe H, Genetic abnormalities in marginal zone B-cell lymphomaHematol Oncol 2000 18(1):01-13. [Google Scholar]