Familial hypercholesterolemia is an autosomal dominant genetic disease characterized by earlier elevated Low-Density Lipoprotein (LDL) cholesterol levels and increased risk for premature Myocardial Infarction (MI). Albeit the diagnosis of some medical Familial Hypercholesterolemia (FH) cases are due to mutations in PCSK9, APOB, or LDLR, detection of mutation rate and profiles relies heavily on different gene pools and ethnicity.
We ran exome sequencing on blood genomic DNA (gDNA) from a 26-year-old Saudi patient on Ion Proton Platform (Ion Torrent, Guilford, Connecticut, USA) as part of a pilot study preluding the establishment of the Saudi Human Genome project. The sequencing results were analysed using Ion suit Bioinformatics system. The patient was matched with a lady of lean body mass and Welsh descent, who suffered from hypercholesterolemia.
The first analysis of known FH genes identified five mutations in APOB, 25 mutations of known genes linked to FH, six mutations in LPR2, one mutation in LDLR, and three mutations in PCSK9. Finally, using disease filter algorithms, we filtered out more than 2000 intronic synonymous variants with likely no biological functions. No major new locus was found in FH. However, via variant reduction and TVC protocols we detected 15 new variants, among which 14 genes are linked to hypercholesterolemia, type-I, and type-II diabetes. We also detected three mutations in PCSK9 and confirmed one by Sanger. Taken together, this report suggests that the genetic determinant of FH in this Saudi patient is likely to be heterogeneous, complicating the diagnostic and novel gene discovery process.
Case Report
During nutritional awareness programs organized by the School of Applied Health Sciences of the University of Hail, we collected random blood samples from 35 individuals. Based on their medical histories, we singled out one patient. This individual is a male Saudi, 26-year-old, who suffered from hypercholesterolemia, diabetes, hypertension, and overweight (BMI 27.5 kg/m2). He is taking lipitor. His family history stated that his younger brother, as well as his father, suffered from FH and all was taking lipitor. We matched him with a 58-year-old female, a Caucasian of Welsh descent. This second individual is of lean body mass and suffered from hypercholesterolemia [Table/Fig-1].
Distribution of plasma lipids and cholesterol among the two subjects.
| Subjects | Ages (years) | Total Cholesterol* | HDL* | LDL* | VLDL* | TG* |
|---|
| 1 | 58 | 7.3 | 0.8 | 5.7 | 3.5 | 1.7 |
| 2 | 26 | 8.4 | 0.6 | 6.1 | 4.1 | 1.9 |
| Normal range | NA | 1-1.3 | 1.3 | 1.42 | 1.5 | 0.7 |
In mmol/L HDL: High-Density Lipoprotein, LDL: Low-Density Lipoprotein, VLDL: Very Low-Density Lipoprotein: triglycerides
Exome library preparation and DNA sequencing [
Table/Fig-2]
Exome sequencing analysis pipeline listing various steps and tools for processing Ion Torrent data.
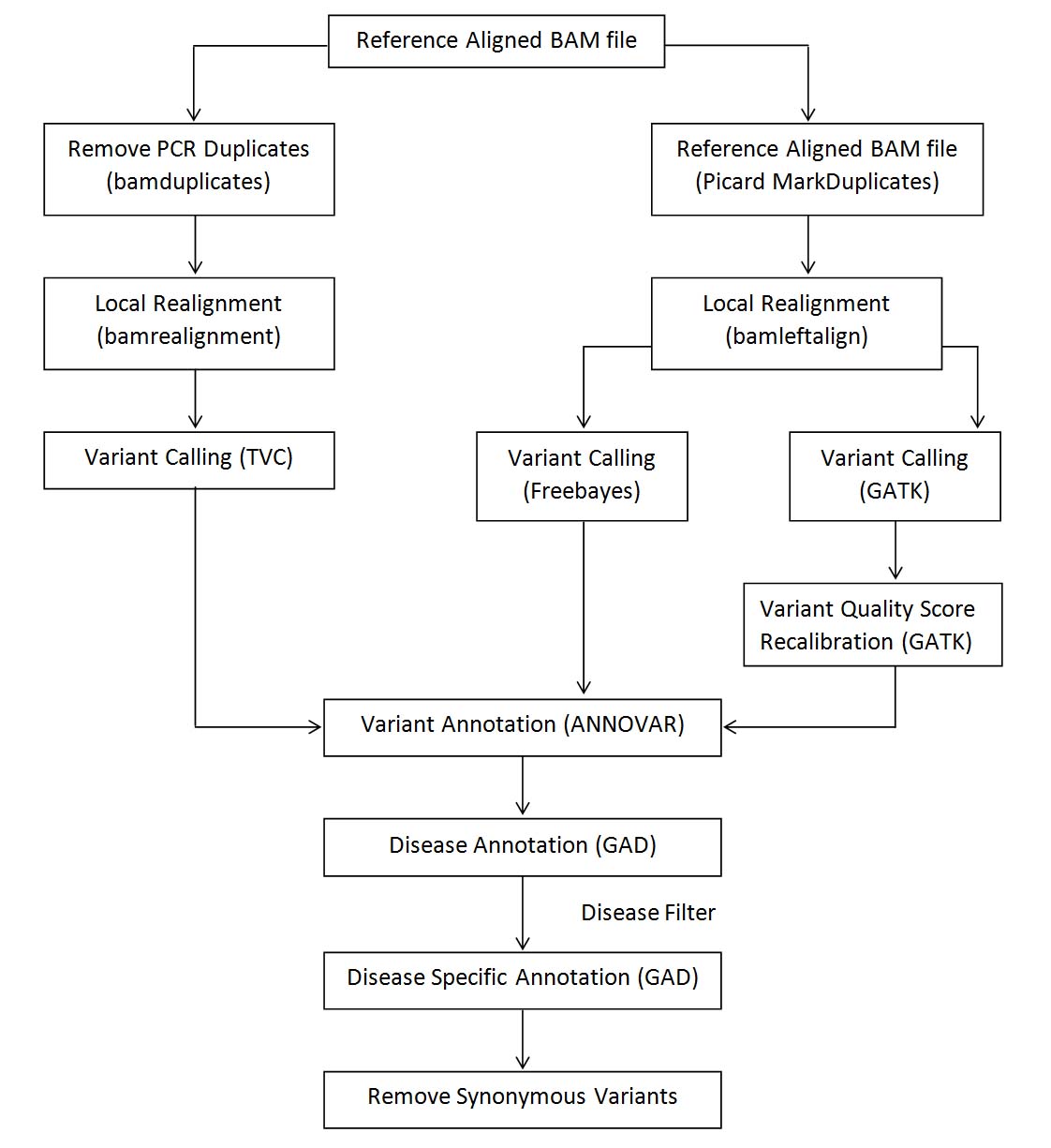
High quality gDNA was purified from whole blood using a Genomic DNA Purification Kit (QIA amp DNA Blood Mini Kit from Qiagen, Hilden, Germany. The library construction was conducted using The Ion AmpliSeq™ Exome RDY Kit 4x2 configuration, which includes the following components for eight exomes (running 2 exomes per Ion PI™ Chip v3):
50 ng of gDNA was utilized using transposase-based kit chemistry to create exome libraries;
The library was quality controlled using the Bioanalyser machine
(Agilent, Maryland);
2-6 pM libraries were used in a 2 x 76 paired end cycles to perform (+1 cycle to each forward and reverse read to allow for phasing/pre-phasing).
Bioinformatics analyses
FastQC files were concatenated from multiple runs;
Adapter trimming and base quality scores (less than Q30) were removed using Cut adapt (V1.7.1);
FastQC (V0.11.2) was used to check primary and post trimmed sequences;
Alignments to the reference human genome (hg19) were conducted using BWA (version 0.7.15);
The Genome Analysis Tool Kit, GATK (version 3.0.0), was used for base quality score recalibration, variant calling following by hard filtering to identify high-quality variants for downstream analyses;
SnpEffv4.1 was exploited to determine in silico impacts upon protein function of candidate genes.
Results and Data Analysis for Sample Variant Reduction and Filtering Results
Using the variant filtering protocol, as illustrated in [Table/Fig-3,4], TVC generated 2250 variants from our patient; 12,800 variants from Freebayes, and 4089 from GATK. Furthermore, annotation and filtering with GAD for the resulting variants generated 11 variants from TVC (corresponding to seven genes) reported in [Table/Fig-5] (Supplementary Material), 60 variants for Freebayes (corresponding to 29 genes) reported in [Table/Fig-6], and 18 variants for GATK (corresponding to 17 genes) reported in [Table/Fig-7].
Variant reduction pipeline performed using ANNOVAR tool.
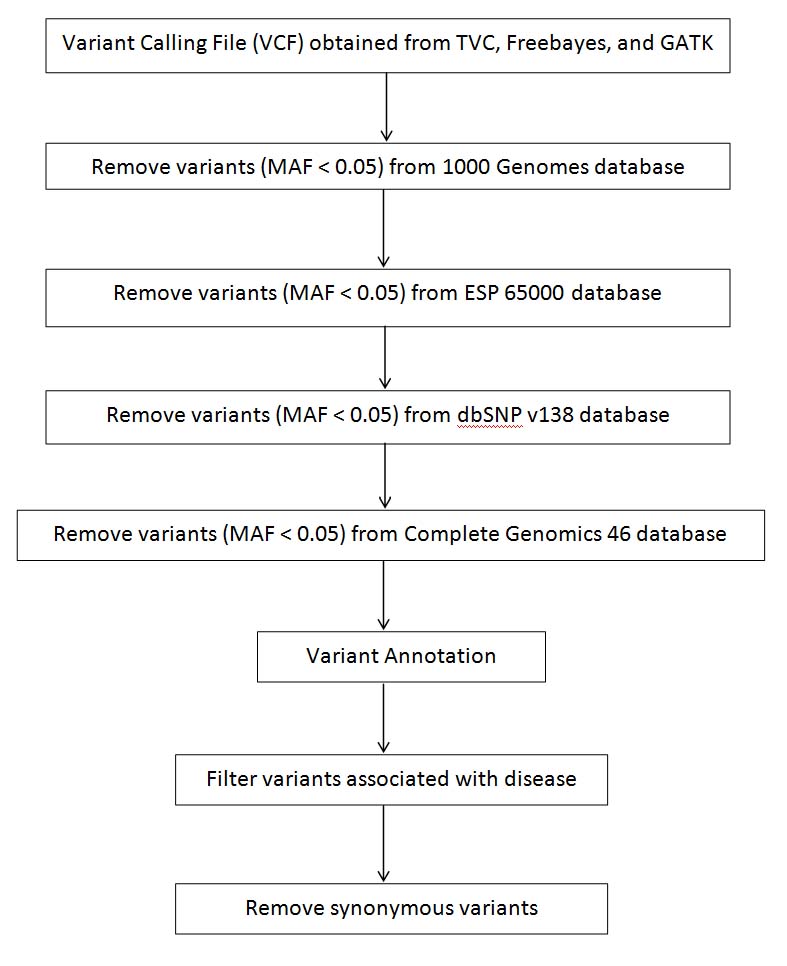
Schematic of the steps illustrating variant reduction protocol performed for Sample-1 and Sample-30. (Please note that variants were filtered having MAF lower than 0.05.).
List of variants obtained from variant reduction protocol associated with hypercholesterolemia in Sample-30 using TVC [1-3].
| Chr | Gene | Start | End | Ref | Alt | Function | Ref |
|---|
| chr17 | ACACA | 35478221 | 35478221 | C | - | intronic | |
| chr12 | ACACB | 109637188 | 109637188 | G | - | intronic | |
| chr2 | INSIG2 | 118847182 | 118847182 | - | T | intronic | |
| chr2 | INSIG2 | 118847183 | 118847183 | G | C | intronic | |
| chr2 | INSIG2 | 118847184 | 118847184 | T | A | intronic | |
| chr2 | INSIG2 | 118847182 | 118847182 | T | - | intronic | |
| chr1 | LDLRAP1 | 25889632 | 25889632 | T | - | exonic | [1] |
| chr15 | LIPC | 58838037 | 58838037 | - | G | exonic | [2] |
| chr2 | LRP2 | 170037992 | 170037992 | G | - | exonic | [3] |
| chr2 | LRP2 | 170037991 | 170037992 | AG | ACG | exonic | [3] |
| chr7 | NPC1L1 | 44571892 | 44571892 | A | - | intronic | |
List of variants obtained from variant reduction protocol associated with hypercholesterolemia in Sample-30 using Freebayes [4-9].
| Chr | Gene | Start | End | Ref | Alt | Function | Ref |
|---|
| chr9 | ABCA1 | 107586838 | 107586838 | G | - | exonic | |
| chr9 | ABCA1 | 107579565 | 107579571 | TGGGGGA | TGGGGA | intronic | |
| chr7 | ABCB1 | 87179703 | 87179710 | CTTTTTTA | CTTTTTTTA | intronic | |
| chr2 | ABCG8 | 44079832 | 44079832 | G | - | exonic | |
| chr17 | ACACA | 35479457 | 35479457 | G | - | exonic | |
| chr17 | ACACA | 35518723 | 35518723 | G | - | exonic | |
| chr17 | ACACA | 35598978 | 35598985 | TAAAAAAG | TAAAAAAAG | intronic | |
| chr17 | ACACA | 35597450 | 35597450 | C | - | exonic | |
| chr17 | ACACA | 35614782 | 35614782 | T | - | exonic | |
| chr12 | ACACB | 109637187 | 109637189 | CGA | CCA | intronic | |
| chr12 | ACACB | 109604630 | 109604630 | C | - | intronic | |
| chr12 | ACACB | 109604774 | 109604782 | TGGGGGGGA | TGGGGGGA | exonic | |
| chr12 | ACACB | 109677804 | 109677811 | AGGGGGGC | AGGGGGC | intronic | |
| chr17 | ACE | 61568535 | 61568541 | ACCCCCA | ACCCCA | intronic | |
| chr17 | ACE | 61566516 | 61566516 | G | T | intronic | |
| chr17 | ACE | 61568448 | 61568448 | G | - | intronic | |
| chr17 | ACE | 61561945 | 61561945 | T | C | intronic | |
| chr17 | ACE | 61560015 | 61560015 | A | - | exonic | [4] |
| chr11 | APOA4 | 116691983 | 116691983 | C | A | exonic | |
| chr11 | APOA4 | 116691848 | 116691848 | G | - | exonic | |
| chr11 | APOA5 | 116661072 | 116661074 | GTC | GC | exonic | |
| chr2 | APOB | 21229253 | 21229253 | G | - | exonic | |
| chr2 | APOB | 21234271 | 21234271 | C | - | exonic | |
| chr2 | APOB | 21242614 | 21242614 | G | - | exonic | |
| chr2 | APOB | 21259995 | 21259995 | G | - | exonic | |
| chr2 | APOB | 21229031 | 21229033 | TGG | GGT | exonic | |
| chr16 | CETP | 57003311 | 57003311 | C | - | exonic | |
| chr16 | CETP | 56997142 | 56997143 | CA | CGA | intronic | |
| chr16 | CYBA | 88709633 | 88709633 | C | - | downstream | |
| chr16 | CYBA | 88713044 | 88713050 | GAAAAAC | GAAAAC | intronic | |
| chr10 | CYP2C19 | 96534904 | 96534904 | C | - | exonic | |
| chr12 | GNB3 | 6952520 | 6952520 | C | - | intronic | |
| chr5 | HMGCR | 74647308 | 74647308 | G | - | exonic | |
| chr5 | HMGCR | 74655080 | 74655080 | G | - | exonic | |
| chr3 | ITIH4 | 52863248 | 52863248 | - | G | exonic | |
| chr19 | LDLR | 11221471 | 11221471 | C | - | intronic | |
| chr1 | LDLRAP1 | 25893265 | 25893266 | TG | TCG | intronic | |
| chr1 | LEPR | 66075722 | 66075724 | GCT | GT | exonic | |
| chr1 | LEPR | 66075946 | 66075952 | GAAAAAG | GAAAAAC | exonic | |
| chr15 | LIPC | 58837909 | 58837911 | CTG | CCG | intronic | |
| chr2 | LRP2 | 169985117 | 169985124 | TAAAAAAT | TAAAAAT | UTR3 | |
| chr2 | LRP2 | 169994045 | 169994045 | A | - | intronic | |
| chr2 | LRP2 | 170053498 | 170053498 | C | - | exonic | [3] |
| chr2 | LRP2 | 170053450 | 170053450 | A | - | exonic | [3] |
| chr2 | LRP2 | 170137020 | 170137020 | G | - | exonic | [3] |
| chr2 | LRP2 | 170103853 | 170103861 | CAAAAAAAT | CAAAAAAAAT | intronic | [3] |
| chr11 | LRP5 | 68192646 | 68192646 | G | - | exonic | [5] |
| chr11 | LRP5 | 68207287 | 68207294 | TGGGGGGC | TGGGGGC | exonic | [5] |
| chr11 | LRP5 | 68153746 | 68153746 | G | - | intronic | |
| chr1 | MTHFR | 11863213 | 11863219 | AGGGGGC | AGGGGC | intronic | |
| chr7 | NPC1L1 | 44578837 | 44578843 | TGGGGGC | TGGGGC | exonic | [6] |
| chr7 | NPC1L1 | 44573332 | 44573332 | G | - | intronic | |
| chr1 | PCSK9 | 55512312 | 55512319 | GCCCCCCG | GCCCCCG | exonic | |
| chr1 | PCSK9 | 55529187 | 55529187 | G | - | exonic | |
| chr1 | PCSK9 | 55523200 | 55523200 | C | - | intronic | |
| chr17 | SREBF1 | 17739987 | 17739993 | GCCCCCT | GCCCCT | intronic | |
| chr17 | SREBF1 | 17719973 | 17719973 | G | - | exonic | [7] |
| chr22 | SREBF2 | 42300855 | 42300855 | G | - | intronic | |
| chr22 | SREBF2 | 42290850 | 42290850 | G | - | exonic | [8] |
| chr9 | TLR4 | 120476639 | 120476639 | C | - | exonic | [9] |
List of variants obtained from variant reduction protocol associated with hypercholesterolemia in Sample-30 using GATK [10-13].
| Chr | Gene | Start | End | Ref | Alt | Function | Ref |
|---|
| chr9 | ABCA1 | 107586837 | 107586837 | G | - | exonic | |
| chr2 | ABCG8 | 44102403 | 44102403 | G | - | exonic | |
| chr17 | ACACA | 35564717 | 35564717 | A | - | intronic | |
| chr12 | ACACB | 109702119 | 109702119 | C | - | exonic | |
| chr12 | ACACB | 109604629 | 109604629 | C | - | intronic | |
| chr11 | APOA4 | 116691847 | 116691847 | G | - | exonic | [10] |
| chr2 | APOB | 21259994 | 21259994 | G | - | exonic | [11] |
| chr16 | CYBA | 88713045 | 88713045 | A | - | intronic | |
| chr15 | CYP1A1 | 75013852 | 75013852 | G | - | intronic | |
| chr12 | GNB3 | 6952519 | 6952519 | C | - | intronic | |
| chr5 | HMGCR | 74655079 | 74655079 | G | - | exonic | [12] |
| chr3 | ITIH4 | 52863246 | 52863246 | - | G | exonic | [13] |
| chr19 | LDLR | 11221470 | 11221470 | C | - | intronic | |
| chr1 | LDLRAP1 | 25889632 | 25889632 | T | - | exonic | [1] |
| chr2 | LRP2 | 169985118 | 169985118 | A | - | UTR3 | [3] |
| chr1 | PCSK9 | 55512313 | 55512313 | C | - | exonic | |
| chr17 | SREBF1 | 17718197 | 17718197 | C | - | exonic | [7] |
| chr22 | SREBF2 | 42300854 | 42300854 | G | - | intronic | |
From the above filtering analysis, 15 genes from our patient were found to be common in Freebayes and GATK variants, which were found to be highly associated with hypercholesterolemia, viz., ABCA1, ABCG8, ACACA, ACACB, APOA4, APOB, CYBA, GNB3, HMGCR, ITIH4, LDLR, LDLRAP1, LRP2, PCSK9, and SREBF1.
Furthermore, causal variants from our patient were also identified, based on the association of genes with atherosclerosis, cholesterol, diabetes, hypercholesterolemia, and obesity. Resulting variants were filtered, which were predicted to have “damaging” impact on the protein function using SIFT and POLYPHEN.
[Table/Fig-8,9 and 10] list the variants obtained from the variant reduction protocol, which are associated with diabetes, hypercholesterolemia, cholesterol, and obesity and predicted to be “Damaging.” Results in [Table/Fig-8,9 and 10] are obtained from TVC, Freebayes, and GATK, respectively. From [Table/Fig-8], five variant (five genes) genes are associated with the diseases. Out of five genes, four (viz., ACAP3, FTCD, HLA-DQA1, and RENBP) are associated with type I diabetes, whereas the NCKAP5 gene is associated with cholesterol formation. On the other hand, Freebayes [Table/Fig-9] gave ten variants (nine genes), of which FTCD, HCFC1, HLA-DQA1, OCA2, and TNXB are associated with type I and II diabetes; ADAM23, IL20RA, and NCKAP5 are associated with cholesterol formation; and TCN2 with atherosclerosis. GATK [Table/Fig-10] gave nine variants (seven genes), of which ACOT11, CPAMD8, FTCD, and JUN are associated with type II diabetes, whereas DOCK6, NCKAP5, and TLE2 are associated with cholesterol formation.
List of disease causal non-synonymous variants obtained from variant reduction protocol using TVC, which are predicted to be damaging in our patient.
| Chr | Gene | Start | End | Ref | Alt | DISEASE |
|---|
| chr1 | ACAP3 | 1233962 | 1233962 | A | G | Type 2 Diabetes| oedema | rosiglitazone |
| chr21 | FTCD | 47574116 | 47574116 | G | T | Type 2 Diabetes| oedema | rosiglitazone |
| chr6 | HLA-DQA1 | 32609800 | 32609800 | C | A | Diabetes mellitus type II| Diabetes Mellitus, Type 2 |
| chr2 | NCKAP5 | 133489586 | 133489586 | C | T | Cholesterol |
| chrX | RENBP | 153209587 | 153209587 | C | A | Type 2 Diabetes| oedema | rosiglitazone |
List of disease causal non-synonymous variants obtained from variant reduction protocol using Freebayes, which are predicted to be damaging in Sample-30.
| Chr | Gene | Start | End | Ref | Alt | DISEASE |
|---|
| chr2 | ADAM23 | 207429779 | 207429779 | A | G | Cholesterol |
| chr21 | FTCD | 47574116 | 47574116 | G | T | Type 2 Diabetes| oedema | rosiglitazone |
| chr21 | FTCD | 47574117 | 47574117 | C | T | Type 2 Diabetes| oedema | rosiglitazone |
| chrX | HCFC1 | 153220581 | 153220581 | G | C | Type 2 Diabetes| oedema | rosiglitazone |
| chr6 | HLA-DQA1 | 32609800 | 32609800 | C | A | Diabetes Mellitus, Insulin-Dependent|Diabetes Mellitus, Type 1 |
| chr6 | IL20RA | 137323444 | 137323444 | C | T | Cholesterol, LDL |
| chr2 | NCKAP5 | 133489586 | 133489586 | C | T | Cholesterol |
| chr15 | OCA2 | 28234812 | 28234812 | T | C | Type 2 Diabetes| oedema | rosiglitazone |
| chr22 | TCN2 | 31013431 | 31013431 | G | T | atherosclerosis |
| chr6 | TNXB | 32064062 | 32064062 | C | T | Diabetes Mellitus, Type 1 |
List of disease causal non-synonymous variants obtained from variant reduction protocol using GATK, which are predicted to be damaging in Sample-1and -30.
| Chr | Gene | Start | End | Ref | Alt | DISEASE |
|---|
| chr1 | ACOT11 | 55070857 | 55070857 | A | G | Type 2 Diabetes| oedema | rosiglitazone |
| chr19 | CPAMD8 | 17015101 | 17015101 | C | G | Type 2 Diabetes| oedema | rosiglitazone |
| chr19 | DOCK6 | 11358800 | 11358800 | G | A | Cholesterol, HDL |
| chr21 | FTCD | 47574116 | 47574116 | G | T | Type 2 Diabetes| oedema | rosiglitazone |
| chr21 | FTCD | 47574117 | 47574117 | C | T | Type 2 Diabetes| oedema | rosiglitazone |
| chr1 | JUN | 59247799 | 59247799 | T | C | Type 2 Diabetes| oedema | rosiglitazone |
| chr1 | JUN | 59247800 | 59247800 | G | C | Type 2 Diabetes| oedema | rosiglitazone |
| chr2 | NCKAP5 | 133489586 | 133489586 | C | T | Cholesterol |
| chr19 | TLE2 | 3005827 | 3005827 | G | T | Cholesterol |
Discussion
Familial hypercholesterolemia (FH (OMIM #143890)) is an autosomal dominant genetic disorder plagued with excessive elevation of Low-Density Lipoprotein Cholesterol (LDL-C) caused, in general, by mutations in three genes: LDLR, APOB, and PCSK9 [14]. A recessive form of FH has also been reported and is characterized by mutations in LDLRAP1 [15].
In Saudi Arabia, no National Registry really exists for FH. However, CHD constitutes the number one killer [16]. The importance of FH in the U.K. recommended the use DNA information for diagnostic purpose [17]. A 93% of patients with FH bear their mutations on the LDLR gene [18]. The APOB variant (c.10580G>A, p. (Arg3527Gln)) accounts for ∼5% of U.K. FH, whereas a gain-of-function mutation in PCSK9 {c.1120G>T, p. (Asp374Tyr)} can be found in roughly 1.7% of FH patients [10,19].
The genes annotation, the SNPs and INDELS characterized in this study are scattered on different chromosomes. Most of them are linked to cardiovascular diseases, hypercholesterolemia, and diabetes [Table/Fig-11]. Our patient was hypercholesterolemic, ascertained by the multiple gene variants we observed in exonic position, as assessed by our bioinformatics suite analysis using TVC, Freebayes, and GATK. These observations were also reported by other authors, which we summarized in [Table/Fig-11]. We also reported, via disease association algorithms, some genes in [Table/Fig-8,9 and 10].
Summary list of variants obtained from variant reduction protocol associated with hypercholesterolemia in Sample-1and -30 using TVC, Freebayes, and GATK.
| Chr | Gene | Start | End | Ref | Alt | Function | Ref |
|---|
| chr1 | LDLRAP1 | 25889632 | 25889632 | T | - | exonic | [1] |
| chr2 | LRP2 | 170037992 | 170037992 | G | - | exonic | [3] |
| chr2 | LRP2 | 170037991 | 170037992 | AG | ACG | exonic | [3] |
| chr9 | ABCA1 | 107586838 | 107586838 | G | - | exonic | |
| chr2 | ABCG8 | 44079832 | 44079832 | G | - | exonic | |
| chr17 | ACE | 61560015 | 61560015 | A | - | exonic | [4] |
| chr2 | LRP2 | 170053498 | 170053498 | C | - | exonic | [3] |
| chr2 | LRP2 | 170053450 | 170053450 | A | - | exonic | [3] |
| chr2 | LRP2 | 170137020 | 170137020 | G | - | exonic | [3] |
| chr11 | LRP5 | 68192646 | 68192646 | G | - | exonic | [5] |
| chr11 | LRP5 | 68207287 | 68207294 | TGG-GGG- GC | TGG-GG- GC | exonic | [5] |
| chr7 | NPC1L1 | 44578837 | 44578843 | TGG-GG- GC | TGG-GGC | exonic | [6] |
| chr1 | PCSK9 | 55512312 | 55512319 | GCC-CCC- CG | GC-CC- CCG | exonic | |
| chr1 | PCSK9 | 55529187 | 55529187 | G | - | exonic | |
| chr17 | SREBF1 | 17719973 | 17719973 | G | - | exonic | [7] |
| chr22 | SREBF2 | 42290850 | 42290850 | G | - | exonic | [8] |
| chr9 | TLR4 | 120476639 | 120476639 | C | - | exonic | [9] |
| chr9 | ABCA1 | 107586837 | 107586837 | G | - | exonic | |
| chr2 | ABCG8 | 44102403 | 44102403 | G | - | exonic | |
| chr11 | APOA4 | 116691847 | 116691847 | G | - | exonic | [10] |
| chr2 | APOB | 21259994 | 21259994 | G | - | exonic | [11] |
| chr5 | HMGCR | 74655079 | 74655079 | G | - | exonic | [12] |
| chr3 | ITIH4 | 52863246 | 52863246 | - | G | exonic | [13] |
| chr1 | LDLRAP1 | 25889632 | 25889632 | T | - | exonic | [1] |
| chr2 | LRP2 | 169985118 | 169985118 | A | - | UTR3 | [3] |
| chr17 | SREBF1 | 17718197 | 17718197 | C | - | exonic | [7] |
Most of these variants deal with type 2 diabetes, cholesterol metabolism, atherosclerosis, and oedema, attributes that are characteristic of our patient from the phenotype point of view. Representative studies from 13,745 U.S. men and women found that high prevalence of hypertension, diabetes, dyslipidemia, and metabolic syndrome substantially rise with increasing body mass index. Our findings in this young Saudi patient, with an association of his metabolic state and a plethora of mutated genes (reported in [Table/Fig-11]) linked to hypercholesterolemia, diabetes, oedema, and hypertension, are nothing new. These authors have demonstrated in exhaustive studies that obesity, hypercholesterolemia, and hypertension coexist in conglomerate.
Aside from canonical genes observed linked to FH, like APOB, LDLR, and HMGCR (which are also characterized in our study), we also observed in [Table/Fig-5,6 and 7] the presence of LDLRAP1 and PCSK9, both located on chromosome1. LDLRAP1 (also known as ARH), whose gene product binds (PTD) domain, interacts with the cytoplasmic tail of the LDL receptor. Mutations in this gene lead to LDL receptor malfunction and cause the disorder of Autosomal Recessive Hypercholesterolemia (ARH).
From this Saudi patient, we observed two types of SNPs in the LDLRAP1 gene: one exonic consisting of the deletion of T, at the position 2588932 [Table/Fig-5,7]; and one intronic consisting of insertion of C, at the position 25893265 [Table/Fig-6]. Whether or not this patient suffers from a form of autosomal recessive FH remains to be clarified by further family-wide genetic studies aimed at developing a family pedigree tree. We identified two variants in our patient on the LDLRAP1 genomic sequence. Taken together in the genetic point of view, our patient presents a heterozygocity form of FH as ascertained by the multiple variants we uncovered in this study.
Our data also points out the identification of multiple variants of the PCSK9 gene, characterized by gain of function as ascertained by the ANNOVAR algorithm [Table/Fig-6,7]. These variants are characterized by the deletion of C, G, C, and C at exonic and intronic positions respectively at {55512312-55512319}, 555291871, 55523200, and 55512313. PCSK9 is an important gene involved in cholesterol metabolism and accounts for 1.7% of FH. Its major function is to down-regulate the circulating cholesterol uptake by the liver. Elevated plasma levels of PCSK9 or increased activity of PCSK9 reduces cholesterol uptake and causes hypercholesterolemia. Conversely, a decrease in plasma levels of PCSK9 or decreased activity of PCSK9 increases cholesterol uptake and causes hypocholesterolemia [20].
In our study, we mapped PCSK9 to chromosome 1 and characterized with two different damageable variants in our Saudi patient. PCSK9 is widely known to be involved in the metabolism of cholesterol [20]. PCSK9’s main known function is to degrade the receptor of LDL.
Our patient harbors two SNPs: one on exon 9 R474I (G-A) and one on exon 12 G670E (GLY-GLU). Both of these two variants on PCSK9 are characterized by Gain of Function (GOF)—deleterious for cholesterol clearance, as they accelerate LDL receptors’ degradation. These have been observed in previous studies as well in patients characterized by a hypercholesterolemic state.
Administration of PCSK9-inhibiting humanized monoclonal antibodies has proven to be a very efficient way of reducing hypercholesterolemia [21]. Studies with humans, including phase III clinical trials have demonstrated that PCSK9 inhibition actually does lower those diseases.
A number of monoclonal antibodies that bind to PCSK9 near the catalytic domain that interacts with the LDLR and hence inhibits the function of PCSK9, are currently in clinical trials. From Amgen, mAb-AMG145 in phase II and III clinical trial levels can lower plasma LDL-C by 43-55% [22]. From RegeneronmAb-REGN727/ SAR236553 in the phase II clinical trial level can lower plasma LDL-C by 40-72% [23].
Subsequent studies we undertook led us to uncover other PCSK9 SNPs characterized by loss of function and a Leucine 11 allelic variant in signal peptide of PCSK9 (Leucine-Zipper stretch), all in exon 1. Interestingly, these two motifs cosegregate together and are linked to people with low plasma cholesterol [24] (data not shown and this will be reported separately).
Conclusion
In summary, we have run the whole exome sequencing on a young Saudi blood genomic DNA using ion proton platform. Analysis identified 15 disease-causing variants in already known FH loci, as well as six previously reported APOB variants. Variants filtration using SIFT and POLYPHEN algorithms identified causal damageable genes associated with atherosclerosis, cholesterol, diabetes, hypercholesterolemia, and obesity. Of importance is the identification of two SNPs in the LDLRAP1 gene and four variants in the PCSK9 gene, among which two are highly damageable, imparting a polygenic characterization for this patient afflicted with FH. The two major PCSK9 variants uncovered constitute an excellent clinical tool to genotype this population and ultimately to design monoclonal antibodies treatments.
*In mmol/L HDL: High-Density Lipoprotein, LDL: Low-Density Lipoprotein, VLDL: Very Low-Density Lipoprotein: triglycerides
[1]. Fellin R, Arca M, Zuliani G, Calandra S, Bertolini S, The history of Autosomal Recessive Hypercholesterolemia (ARH). From clinical observations to gene identificationGene 2015 555:23-32. [Google Scholar]
[2]. Kondkar AA, Nair KG, Ashavaid TF, Genetic analysis of Indian subjects with clinical features of possible type IIa hypercholesterolemiaJCLA 2007 21(6):375-81. [Google Scholar]
[3]. Corella D, Ordovas JM, Single nucleotide polymorphisms that influence lipid metabolism:interaction with dietary factorsAnnu Rev Nutr 2005 25:341-90. [Google Scholar]
[4]. Dhar S, Ray S, Dutta A, Sengupta B, Chakrabarti S, Polymorphism of ACE gene as the genetic predisposition of coronary artery disease in Eastern IndiaIndian Heart Journal 2012 64:576-81. [Google Scholar]
[5]. Saarinen A, Saukkonen T, Kivelä T, Lahtinen U, Laine C, Somer M, Low density lipoprotein receptor-related protein 5 (LRP5) mutations and osteoporosis, impaired glucose metabolism and ypercholesterolemiaClin Endocrinol (Oxf) 2010 72(4):481-88. [Google Scholar]
[6]. Villa F, Mutations in NPC1L1 and coronary heart diseaseN Engl J Med 2015 372(9):882-83. [Google Scholar]
[7]. `Laaksonen R, Thelen KM, Paiv H, Matinheikki J, Vesalainen R, Janatuinen T, Genetic variant of the SREBF-1 gene is significantly related to cholesterol synthesis in manAtherosclerosis 2006 185:206-09. [Google Scholar]
[8]. Kim Y, Nam YJ, Lee C, Analysis of the SREBF2 gene as a genetic risk factor for vascular dementiaAJMG 2005 1:19-22. [Google Scholar]
[9]. Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Toll-like Receptor 4 Polymorphisms and AtherogenesisN Engl J Med 2002 343:185-92. [Google Scholar]
[10]. Kamboh MI, Friedlaender JS, Ahn YI, Ferrell RE, A common deletion polymorphism in the apolipoprotein a4 gene and its significance in lipid metabolismArterioscler Thromb 1994 14:656-62. [Google Scholar]
[11]. Tremblay AJ, Lamarche B, Ruel IL, Hogue JC, Bergeron J, Gagné C, Increased production of VLDL apoB-100 in subjects with familial hypercholesterolemia carrying the same null LDL receptor gene mutationJournal of Lipid Research 2004 45:866-72. [Google Scholar]
[12]. Sun C, Huang F, Liu X, Xiao X, Yang M, Hu G, MiR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCRInternational Journal of Molecular Medicine 2015 35:847-53. [Google Scholar]
[13]. Fujita Y, Ezura Y, Emi M, Takada KSD, Iino Y, Takahashi YKK, Hypercholesterolemia associated with splice-junction variation of inter-a-trypsin inhibitor heavy chain 4 (ITIH4) geneJ Hum Genet 2004 49:24-28. [Google Scholar]
[14]. Marks D, Thorogood M, Neil HA, Humphries SE, A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemiaAtherosclerosis 2003 168:1-14. [Google Scholar]
[15]. Ciccarese M, Pacifico A, Tonolo G, Pintus P, Nikoshkov A, Zuliani G, A new locus for autosomal recessive hypercholesterolemia maps to human chromosome 15q25-q26Am J Hum Genet 2000 66:453-60. [Google Scholar]
[16]. El-Hazmi MA, Warsy AS, Prevalence of obesity in the Saudi populationAnn Saudi Med 1997 17(3):302-06. [Google Scholar]
[17]. Wierzbicki AS, Humphries SE, Minhas R, Familial hypercholesterolaemia:summary of NICE guidanceBMJ 2008 337:a1095 [Google Scholar]
[18]. Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, Low-density lipoprotein receptor gene familial hypercholesterolemia variant database:update and pathological assessmentAnn Hum Genet 2012 76:387-401. [Google Scholar]
[19]. Graham CA, McIlhatton BP, Kirk CW, Beattie ED, Lyttle K, Hart P, Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rateAtherosclerosis 2005 182:331-40. [Google Scholar]
[20]. Mbikay M, Mayne J, Chretien M, Proprotein convertases subtilisin/kexin type 9, an enzyme turned escort protein:hepatic and extra hepatic functionsJournal of Diabetes 2013 5:391-405. [Google Scholar]
[21]. Zhang XL, Zhu QQ, Zhu L, Chen JZ, Chen QH, Li GN, Safety and efficacy of anti-PCSK9 antibodies:a meta-analysis of 25 randomized, controlled trialsBMC Medicine 2015 13:123 [Google Scholar]
[22]. Sullivan D, Olsson AG, Scott R, Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients:the gauss randomized trialJAMA 2012 81:1-10. [Google Scholar]
[23]. McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA, Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/ kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapyJ Am Coll Cardiol 2012 59:2344-53. [Google Scholar]
[24]. Pisciotta L, Sallo R, Rabacchi C, Wunsch A, Calandra S, Bertolini S, Leucine 10 allelic variant in signal peptide of PCSK9 increases the LDL cholesterol-lowering effect of statins in patients with familial hypercholesterolaemiaNutrition, Metabolism, and Cardiovascular Diseases 2012 22:831-35. [Google Scholar]