Co-Inheritance of Haemoglobin D-Punjab and Beta Thalassemia - A Rare Variant
Kalyan Mansukhbhai Shekhda1, Alpa C Leuva2, Jyoti G Mannari3, Aashka Vikas Ponda4, Amee Amin5
1 Assistant Professor, Department of Medicine, Pramukhswami Medical College and Research Institute, Karamsad, Gujarat, India.
2 Professor, Department of Medicine, Pramukhswami Medical College and Research Institute, Karamsad, Gujarat, India.
3 Professor and Head, Department of Medicine, Pramukhswami Medical College and Research Institute, Karamsad, Gujarat, India.
4 Postgraduate Trainee, Department of Medicine, Pramukhswami Medical College and Research Institute, Karamsad, Gujarat, India.
5 Intern, Department of Medicine, Pramukhswami Medical College and Research Institute, Karamsad, Gujarat, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Kalyan Mansukhbhai Shekhda, 218, New Shakti Vijay Soc., opposite Surat Supar Store, Hirabaug, Varachha Road-395006, Surat, Gujarat, India.
E-mail: kalokly@gmail.com
Haemoglobinopathies are a frequent cause of anaemia in Northwestern India due to traditional practices of consanguineous marriages. Haemoglobin D-Punjab is one of the most common subvariants (55%) of haemoglobin D, which can be inherited as a homozygous or a heterozygous trait with other haemoglobinopathies. Though, haemoglobin D-Punjab is commonly seen, a heterozygous trait with beta thalassemia is a very rare presentation. Here, we present a rare case of co-inheritance of haemoglobin D-Punjab and beta thalassemia in a 19-year-old male of Indian origin. He came with gradually progressive generalised weakness with easy fatigability for the past two months. No history of similar complaints in the past. On examination, he was pale and icteric with splenomegaly and Grade I hemorrhoids on systemic examination. On investigation, there was severe anaemia, pancytopenia (mixed picture on smear), vitamin B12 deficiency and raised Lactate Dehydrogenase (LDH). Haemoglobin electrophoresis showed co-inheritance of haemoglobin D-Punjab and beta thalassemia. After Pack Cell Volume (PCV) and B12 supplements, haemoglobin improved. He was counseled about his disease and advised regular follow-up.
Anaemia, Haemoglobinopathies, Lactate dehydrogenase, Splenomegaly
Case Report
A 19-year-old male patient of Indian origin presented with a history of gradually progressive generalised weakness and easy fatigability for past two months. He also had complaints of fresh blood in stool for last two months, accompanied by pain during defecation.
No past history of similar complaints, tuberculosis, significant weight loss, malignancy, or worms in stool could be elicited. The patient was a non-smoker, non-alcoholic with no known history of drug abuse. No similar history could be elicited in family members.
On examination, the patient was conscious, cooperative and oriented to time, place and person. The patient was vitally stable with the pulse of 80 beats per minute and blood pressure of 110/70 mmHg. Pallor and icterus were present on general examination. However, there was no oedema, cyanosis, clubbing and lymphadenopathy. On systemic examination, spleen was palpable just below left costal margin. Per rectal examination revealed Grade 1 hemorrhoids. All other systemic examinations were unremarkable.
Laboratory investigations revealed: Haemoglobin 2.5 g/dl, haematocrit 7.3%, Red Blood Cell (RBC) counts 1.08 million cells/microliter (µl), White Blood Cell (WBC) counts 1800 cells/µl, platelet counts 45000 cells/µl, Mean Corpuscular Volume (MCV) 67.6 fl, Mean Corpuscular Haemoglobin (MCH) 23.1 pg, Mean Corpuscular Haemoglobin Concentration (MCHC) 34.2%, corrected reticulocyte count 0.2%. Peripheral smear was suggestive of a severe microcytic hypochromic picture with few normochromic normocytic RBCs with tear drop cells, elliptocytes and fragmented RBCs. Few target cells and occasional macrocytes were seen. LDH level was 1986 U/L, vitamin B12 level was 51 mcg which was significantly low. Bilirubin levels were: Total (T): 3.5 mg/dl, Indirect (I): 3.31 mg/dl, Direct (D): 0.19 mg/dl. Protein levels and liver enzymes were within normal ranges ruling out any hepatic pathology. Creatinine was 0.61 mg/dl. Direct and indirect Coomb’s tests were negative ruling out any autoimmune causes. Sickling test was negative, thereby ruling out sickle cell anaemia which is quite prevalent in the area.
Routine microscopy of urine was normal, while routine microscopy of stool was negative for worms but fresh blood was found in stool. Ultrasonography of abdomen and pelvis revealed an enlarged spleen of size 14.5 cm with normal liver size.
After clinical examination and routine investigations, some sort of haemoglobinopathy was considered as one of the differential diagnosis as it is very common but underdiagnosed in this area. HB electrophoresis was ordered to rule out haemoglobinopathies. HB electrophoresis revealed findings presented in [Table/Fig-1].
HB electrophoresis report of the patient.
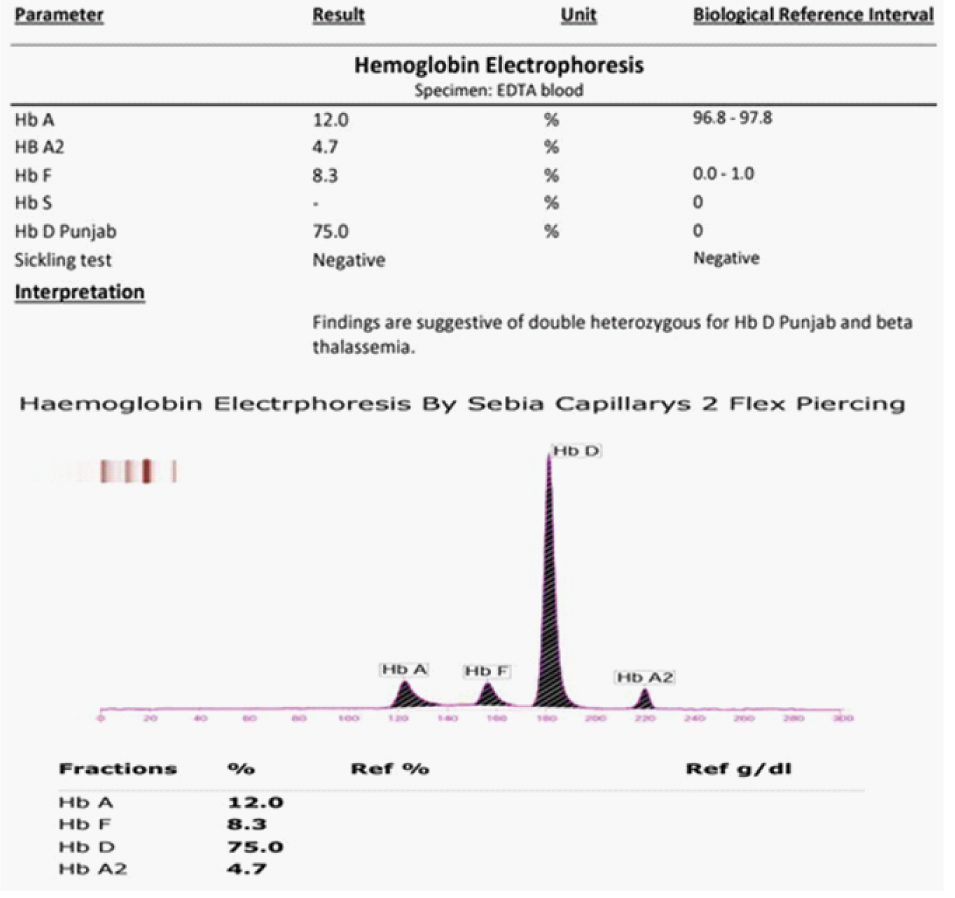
Therapeutically, four Packed Cell Volume (PCV) were transfused over 48 hours. Intravenous (IV) B12 was given daily in a dose of 1500 mcg and was continued in oral form for 15 days after initial five IV doses. Hemorrhoids were treated with lignocaine local application, laxative and Sitz bath. Corrected reticulocyte count on day 4 was 1.9%. The patient was counseled regarding his diagnosis and was discharged on day 6 with medication of B12 deficiency. Regular follow-up was advised and after 12 days, repeat investigations showed an improvement in the clinical profile as depicted in [Table/Fig-2].
Trend of blood indices of the patient since the day of admission.
| Day 1 | Day 2 | Day 4 | Day 5 | Day 18 |
|---|
| Total Count (×1000/μl) | 1.8 | 1.2 | 2.2 | 2.5 | 12.6 |
| Platelet (cells/μl) | 45000 | 60000 | 25000 | 20000 | 7,98,000 |
| Hb (gm/dl) | 2.5 | 5 | 7.4 | 7.6 | 11.5 |
| RBC count (millions/μl) | 1.08 | 1.88 | 3.05 | 3.06 | 4.93 |
| MCV (fl) | 67.6 | 77.7 | 75.4 | 75.5 | 73.6 |
| LDH (U/L) | 1986 | - | - | 887 | - |
| Retic (%) | 0.2 | - | - | 1.9 | 1.6 |
| Vit B12 (mcg) | 51 | - | - | - | - |
| Bilirubin(D) (mg/dl)(I) (mg/dl) | 0.193.31 | 0.232.77 | 0.201.2 | 0.361.14 | - |
Discussion
Haemoglobin D-Punjab is one of the most common subvariants (55%) of haemoglobin D which is a variant of normal haemoglobin derived from a point mutation in the beta globin gene. There is a substitution of glutamine for glutamic acid in the first base of the 121 codon of the beta globin chain. Haemoglobin D-Punjab is quite prevalent in Northwestern India, specifically in Punjab and Gujarat regions communities such as Sikhs, Lohana and Sindhis due to cultural practices of consanguineous marriages [1]. It is also commonly seen in Italy, Belgium, Austria, Turkey (57.9%) and China (55.6%). It can be inherited as a homozygous component or as a heterozygous trait with normal Hb-A. The trait presents with no clinical or haematological alterations. However, when co-inherited with another variant of haemoglobin such as sickle cell or thalassemia, it may present with clinically significant conditions often requiring hospital admissions and blood transfusions [1].
Studies conducted previously have demonstrated that co-inheritance of beta thalassemia with haemoglobin D-Punjab has shown a clinical picture of chronic hemolytic anaemia of moderate severity as well as an association with haematological malignancies [1]. A hypochromic, microcytic red cell picture with reduced red cell indices was reported by Pandey S et al., along with clinically symptomatic patients for moderate anaemia [1]. In a review of case series, three heterozygous individuals for haemoglobin D-Punjab and beta thalassemia were followed out of which only one case had a diagnosis based on clinical symptoms. The other two were found in routine prenatal screening further demonstrating the variability in the clinical picture of coinheritance of haemoglobin D-Punjab with beta thalassemia [2,3]. In our case, the patient had no previous history of any hospital admissions or any transfusions in 19 years of his life. Even with a severely low Hb of 2.5 g/dl, the patient had no symptoms before two months of presentation. While such a delayed visit to the hospital may be attributed to patient’s low socio-economic status and lack of awareness, it is still surprising that the patient felt no discomfort in the initial clinical stages of anaemia. This may be explained by body’s ability to adapt to the chronic state of anaemia. Another possibility for such a late presentation could be an acute drop in the haemoglobin attributed to the fresh blood found in stool as a result of hemorrhoids. While the patient may have been in a clinically moderate state of anaemia, the hemorrhoids since two months may have escalated the drop in the haemoglobin. During follow-up visits, surgical reference for haemorrhoids was taken, and the patient was cleared for no more fresh blood in the stool. An improvement in the blood indices was also seen, providing evidence that no acute blood loss was present [Table/Fig-2].
While the gold standard test for diagnosis of haemoglobinopathies is molecular studies, most cases in this region are diagnosed based on High-Performance Liquid Chromatography (HPLC) and Capillary Electrophoresis (CE) which are more affordable in this low-income population [4]. The principle of HPLC and CE is based on the difference in electrophoretic mobility of different HB variants. Normally, under alkaline pH, electrophoretic mobility of haemoglobin D-Punjab is same as haemoglobin S; however, in acidic pH, its mobility is similar to haemoglobin A [5]. In a normal person, level of haemoglobin A as per CE is between 96%-98% with haemoglobin F of less than 1% and haemoglobin A2 of less than 2% [6]. In our case, as we separated all haemoglobin molecules and variants, we found very high percentages of haemoglobin D-Punjab and co-inheritance of beta thalassemia trait with levels of HbA2 of 4.7% [Table/Fig-1]. Lastly, molecular studies are always recommended as a final investigation, but not followed through with in most cases.
Conclusion
With only a few case reports being available on co-inheritance of haemoglobin D haemoglobinopathies with alpha or beta mutations, a lack of clarity exists with regards to the presence and severity of clinical features. Further standardised studies using gold standard methods and detailed clinical assessments in Northwestern Indian population could provide a larger cohort for gaining a more accurate clinical picture of this co-inheritance. Haemoglobin D-Punjab and beta thalassemia trait co-inheritance being a rare entity and prevailing in north-western India should also be considered in patients of Gujarat presenting as chronic anaemia.
[1]. Pandey S, Mishra RM, Pandey S, Shah V, Saxena R, Molecular characterization of haemoglobin D-Punjab traits and clinical-hematological profile of the patientsSao Paulo Med J 2012 130(4):248-51. [Google Scholar]
[2]. Das S, Mashon RS, Coinheritance of Hb D-Punjab and beta-thalassemia: diagnosis and implications in prenatal diagnosisHemoglobin 2015 39(2):138-40. [Google Scholar]
[3]. Bhat VS, Mandal AK, Mathew B, Co-inheritance of HBD Iran/beta thalassemia IVS1-5 (G >C) Trait in a Punjabi lady with diabetesIndian Journal of Clinical Biochemistry 2012 27(2):202-206. [Google Scholar]
[4]. Sachdev R, Dam AR, Tyagi G, Detection of Hb variants and hemoglobinopathies in Indian population using HPLC: report of 2600 casesIndian Journal of Pathology and Microbiology 2010 53(1):57-62. [Google Scholar]
[5]. Torres L, de S, Okumura JV, Silva DGH, da Bonini-Domingos CR, Hemoglobin D-Punjab: origin, distribution and laboratory diagnosisRevista Brasileira de Hematologia e Hemoterapia 2015 37(2):120-26. [Google Scholar]
[6]. Hafiza A, Malisa MY, Khirotdin RDA, Azlin I, Azma RZ, Chong MKT, HbA2 levels in normal, β-thalassaemia and haemoglobin E carriers by capillary electrophoresisMalaysian J Pathol 2012 34(2):161-64. [Google Scholar]