Alexander Disease (AD) is an autosomal dominant leukodystrophy and occurs predominantly in infants and children. It usually results in death within ten years after onset. Among the four subtypes, infantile form comprises the most of affected individuals. It presents in the first two years of life, typically with progressive psychomotor deficiency, loss of developmental milestones, seizures, and pyramidal signs. Clinical and magnetic resonance image findings usually establish diagnosis of AD. Here, we present a case of Infantile AD with characteristic clinical and radiological features.
Leukodystrophy, Magnetic resonance imaging, Seizures
Case Report
A five-year-old boy reported to the Department of Oral Medicine and Radiology, with painful decayed tooth in the upper right back jaw region for 10 days. The pain was mild, continuous, non-radiating in nature with no associated factors. The patient’s parents gave history of delayed developmental milestones of their son. There was no history of consanguineous marriage of the patient’s parents. They reported their son to be normal at birth with standard birth weight and head circumference. There was history of seizures six months after birth, with gradual psychomotor deficiencies and cognitive abnormalities, for which his parents sought paediatric consultation immediately at seven months of age. Initial consultation was inconclusive due to lack of diagnostic evidence and financial constrictions. However, he was put on phenytoin oral suspension twice daily, which controlled the symptoms. The mother also complained of gradually increasing feeding difficulties in the later months, accompanied by episodes of vomiting. Subsequently, few non-contributory medical investigations were done in the later years. Patient’s family history was found to be insignificant.
On general physical examination, gait and postural abnormality was noted (he could only walk few steps without support). The patient had discrepancy in understanding, inability to speak and respond and presented with social skill deficiency. The head circumference was found to be within normal limits but the occipital region appeared to be flattened. On intraoral examination, 54 and 55 were found to be grossly carious with hypoplastic teeth seen in the posterior region.
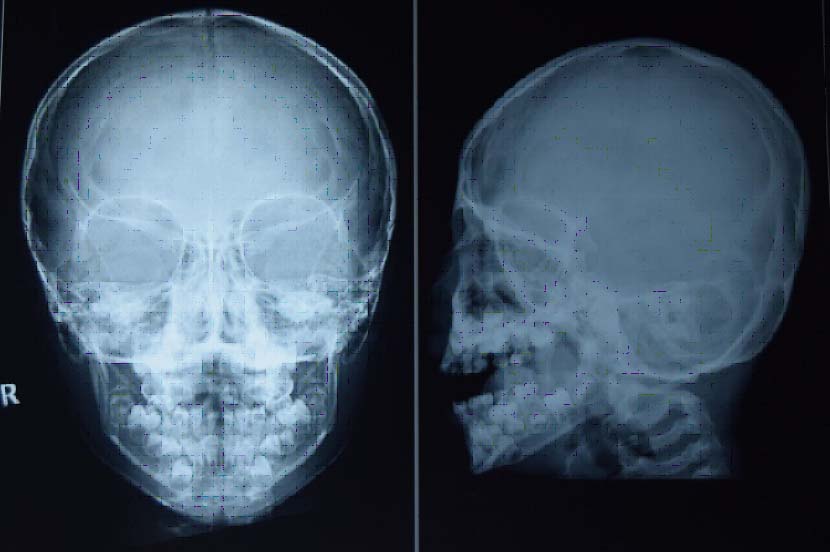
The patient had to undergo extraction regarding the same. Henceforth, pre-anaesthetic evaluation was necessary and he was subjected to various haematological and radiological investigations. Haematological parameters were found to be normal. Postero-anterior view of skull showed no evidence of sutural defects. Lateral skull view revealed flattened occiput [Table/Fig-1].
Postero-anterior view of skull showing no evidence of sutural defects and lateral skull view showing flattened occiput.
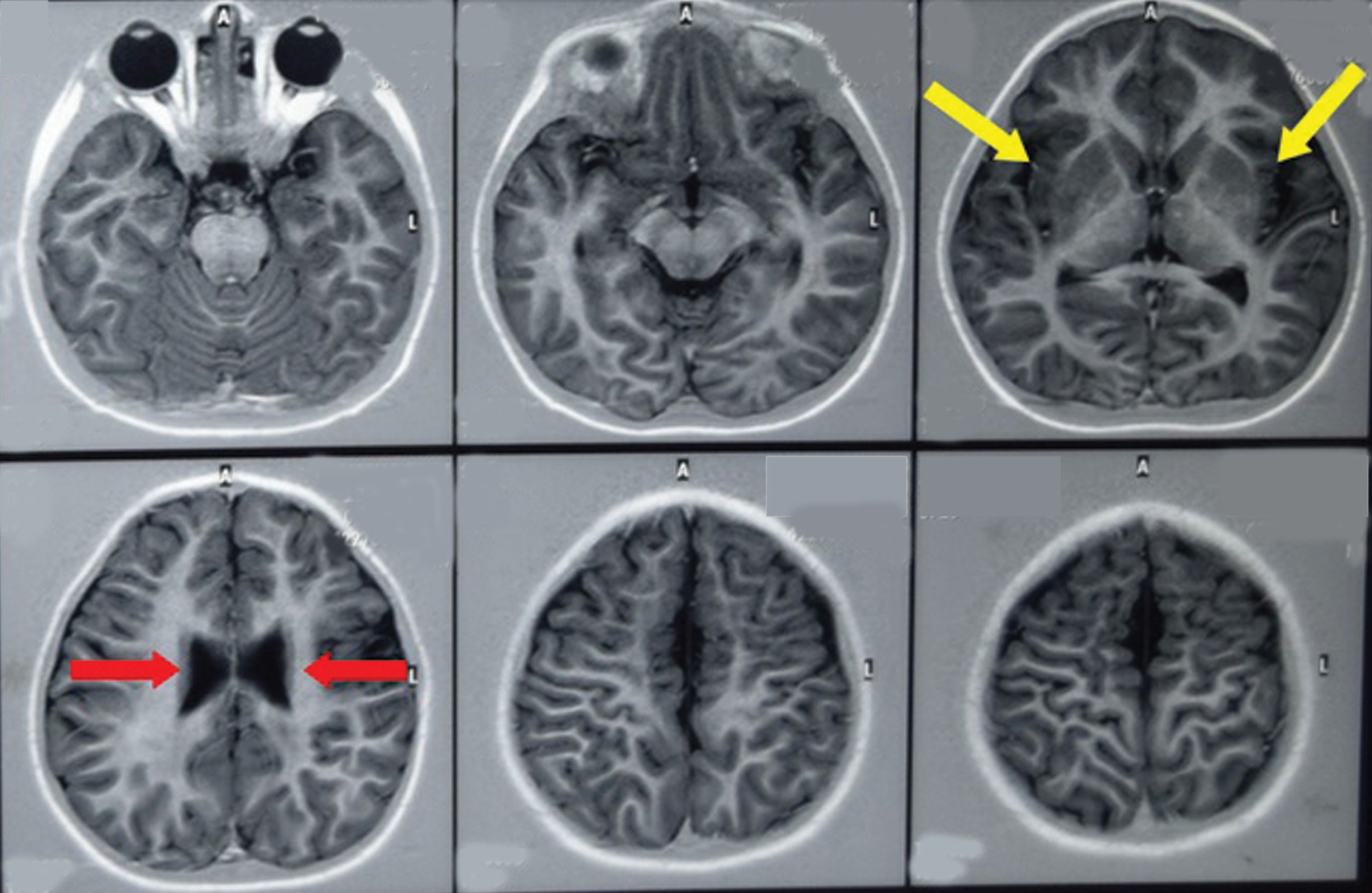
MRI revealed extensive white matter abnormalities, reduced white matter intensity in both frontal lobes with prominent parafalcine and sulcal region, thinning of body of corpus callosum and presence of periventricular rim of decreased signal intensity on T2 weighted images suggestive of bilateral centrum semiovale and left fronto-parietal signal intensity changes with bilateral frontal lobe demyelination, left para-syvliangliotic changes and mid corpus callosal dysgenesis [Table/Fig-2]. Therefore, based on the clinical and radiological presentation, a final diagnosis of Infantile AD was established.
MRI showing extensive white matter abnormalities, reduced white matter intensity in both frontal lobes with prominent parafalcine and sulcal region, thinning of body of corpus callosum (red arrows) and presence of periventricular rim of decreased signal intensity on T2 weighted images (yellow arrows).
In regards to dental treatment, 54 and 55 were being extracted under local anaesthesia and no postoperative complications were reported. The patient’s parents were counselled and advised to consult their paediatrician for cognitive and behavioural therapy.
Discussion
AD is one of the leukodystrophies cornered down to dominant missense mutation involving Glial Fibrillary Acidic Protein (GFAP) gene [1,2]. According to the presenting age, AD is classified into four types: neonatal, infantile, juvenile and adult onset, of which infantile type constitutes more than half of diagnosed cases [3,4].
Onset of Infantile AD is usually within two years of life and often presents with seizures, developmental delays, spastic paresis, psychomotor deficiencies, macrocephaly and progressively grave clinical course [5,6]. Pathological hallmark of disease is presence of rosenthal fibres all over CNS. Diagnosis of AD is usually established by clinical and MRI findings [3,6].
Stewart Alexander in 1949 first described this form of leukodystrophy as progressive degeneration of fibrillary astrocytes, since then several subtypes have been recognized [3]. Neonatal form is usually recognized after birth, infantile form within two years of age, juvenile form within four to fourteen years of age and adult after eighteen years of age. Significantly Infantile AD presents with seizures, developmental delays, spastic paresis, psychomotor deficiencies, macrocephaly and progressively grave clinical course [3,4,6].
In the present case, birth weight and head circumference at birth was normal. The child attended smile and sound response and head holding capacity within four months after birth. Events of seizures started six months after birth which was accompanied by progressive generalized weakness, psychomotor deficiencies, seizures, and developmental delays. The head circumference was within normal range but the occipital region appeared to be flat.
Most literatures reporting Infantile AD presents with macrocephaly as one the important clinical feature, the head circumference of the case was within normal range. Infantile AD with normal head circumference is also reported by Rodriguez D et al., and Goyal M et al., [7,8].
In the current case diagnosis was made by the criteria established by van der Knaap MS et al., as the MRI revealed extensive white matter abnormalities, reduced white matter intensity in both frontal lobes with prominent parafalcine and sulcal region, thinning of body of corpus callosum and presence of periventricular rim of decreased signal intensity on T2 weighted images [9,10]. MRI is preferred over histopathological as presence of rosenthal fibre is also encountered in multiple sclerosis, craniopharyngioma, arteriovenous malformations, neoplasms (pilocytic astrocytoma, ganglioglioma, pleomorphic xanthoastrocytoma) and drug toxicity (quinolones).
The differential diagnosis of AD include disorders that present with macrocephaly and/or cerebral white matter changes; adrenoleukodystrophy, Canavan disease, megalencephalic leukoencephalopathy with subcortical cysts, metachromatic leukodystrophy and multiple sclerosis [11]. Genetic evaluation was not performed in the present case because conclusive diagnosis could be reached by means of MRI.
Oral health care providers play an imperative role in case of Infantile AD. Children often present with dental caries, teeth hypoplasia, gingival and periodontal pathologies, medication-induced gingival hyperplasia, and delayed healing. Episodes of seizures in the backdrop of AD may also cause traumatic injuries in the tongue, buccal and labial mucosa, resulting in bleeding. Parents should bring the affected child to the pedodontist for immediate intervention and treatment. They should be educated about the medical condition and informed about the treatment plan [12]. Mouth-guards should be recommended in children with uncontrolled epilepsy. Pedodontists should discourage the consumption of cariogenic foods and beverages, encourage topical fluorides and sealant application, recommend sugar-free medication, daily use of fluoridated toothpastes and maintain proper oral hygiene [1,12].
Conclusion
AD is a myelin degenerative disorder presenting with seizures, spastic paresis, progressive psychomotor deficiency and grave clinical consequence. Oral health care providers are not ancillaries, but play an important role in providing basic support to the patients. The current discusses the classical clinico-radiological findings of AD, and importance of oral health care providers in management of the same.
[1]. Kohlschütter A, Eichler F, Childhood leukodystrophies: a clinical perspectiveExpert Rev Neurother 2011 11(10):1485-96. [Google Scholar]
[2]. Nishibayashi F, Kawashima M, Katada Y, Murakami N, Nozaki M, Infantile-onset Alexander disease in a child with long-term follow-up by serial magnetic resonance imaging: a case reportJournal of Medical Case Reports 2013 7:194 [Google Scholar]
[3]. Ashrafi MR, Tavasoli A, Aryani O, HoomanAlizadeh H, Houshmand M, Alexander disease: report of two unrelated infantile form cases, identified by GFAP mutation analysis and review of literature;the first report from IranIran J Pediatr 2013 23(4):481-84. [Google Scholar]
[4]. Vazquez E, Macaya A, Mayolas N, Arevalo S, Poca MA, Enriquez G, Neonatal Alexander Disease: MR imaging prenatal diagnosisAm J Neuroradiol 2008 29:1973-75. [Google Scholar]
[5]. Singh N, Bixby C, Etienne D, Tubbs RS, Loukas M, Alexander’s disease: reassessment of a neonatal formChilds Nerv Syst 2012 28:2029-31. [Google Scholar]
[6]. Kumar KJ, Suryaprakash H, Manjunath VG, Harsha S, Infantile Alexander disease: A rare leukodystrophyJ Pediatr Neurosci 2012 7(2):117-19. [Google Scholar]
[7]. Rodriguez D, Gauthier F, Bertini E, Bugiani M, Brenner M, N’guyen S, Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlationAm J Hum Genet 2001 69(5):1134-40. [Google Scholar]
[8]. Goyal M, Mehndiratta S, Faruq M, Dwivedi MK, Kapoor S, Infantile onset alexander disease with normal head circumference: a genetically proven case reportJournal of Clinical and Diagnostic Research 2014 8(11):PD03-PD04. [Google Scholar]
[9]. van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Alexander disease: diagnosis with MR imagingAm J Neuroradiol 2001 22:541-52. [Google Scholar]
[10]. Jung YJ, Jeon BS, The first Korean case of adult-onset Alexander DiseaseNeurology Asia 2014 19(2):207-09. [Google Scholar]
[11]. Wippold FJ, IIPerry A, Lennerz J, Neuropathology for the neuroradiologist: rosenthal fibersAm J Neuroradiol 2006 27:958-61. [Google Scholar]
[12]. Aragon CE, Burneo JG, Understanding the patient with epilepsy and seizures in the dental practiceJ Can Dent Assoc 2007 73(1):71-76. [Google Scholar]