Haemolytic anaemia is the anaemia which results from increased rate of red cell destruction. These anaemia are a heterozygous group of disorders which can be broadly classified as those due to intra corpuscular defects in the RBC and those due to extra corpuscular defects [1]. The haemolytic anaemia resulting from intra corpuscular defects are predominantly hereditary in nature. These defects in the RBCs could be in the haemoglobin molecule (e.g., thalassaemia, sickle cell anaemia), in the red cell membrane (e.g., hereditary spherocytosis, hereditary elliptocytosis) and in the red cell enzymes (e.g., G6PD deficiency and pyruvate kinase deficiency). These hereditary haemolytic anaemia are a cause of significant morbidity and mortality worldwide, placing a large burden on the patients, their families and ultimately on the communities. They can be prevented by population screening, genetic counselling and prenatal diagnosis [1].
These disorders are prevalent in certain communities. Few earlier studies have identified that particular abnormal gene is more prevalent in particular region like beta thalassaemia in North-West and Far East (Sindhis, Gujaratis, Bengalis, Punjabis and Muslims) [2], alpha thalassaemia in tribal population of Andhra Pradesh and Gujarat [3], Sickle cell disease predominately in central India (Madhya Pradesh, Orissa, Andhra Pradesh, Gujarat and to lesser extent in Tamil Nadu, Karnataka, Kerala and Uttar Pradesh) [4].
Hence, a comprehensive hospital based study was carried out on all the hereditary haemolytic anaemia patients reported in the referral hospital between 1st July 2003 to 30th December 2008 and clinical and haematological parameters were correlated.
Materials and Methods
Source of Data
Retrospective study: This group included all patients diagnosed of hereditary haemolytic anaemia based on peripheral smear, special haematological tests like sickle cell preparation, osmotic fragility test before and after incubation, methaemoglobin reduction test, G6PD assay and haemoglobin alkaline electrophoresis from 1st July 2003 to 30th June 2007. The case records of the patients were retrieved from the medical records section of the hospital. The clinical and haematological parameters were recorded.
Prospective study: This group included all cases of hereditary haemolytic anaemia seen in outpatient and inpatient basis from 1st July 2007 to 30th December 2008. The data was analysed similar to that of retrospective study.
Ethical clearance was obtained from the institutional ethical board.
Methods
The study included all the patients with hereditary haemolytic anaemias. Cases where medical records were not available and cases with incomplete clinical details were excluded from the study.
Haematological parameters like cell counts (Total WBC counts, RBC count), red cell indices (Hb, PCV, MCV, MCH, MCHC, RDW) were analysed on automated haematology analyser (sysmex xt1800 and xt2000i). Peripheral smears were stained with Leishman stain to assess the morphology of RBCs.
Sickle cell preparation was done on a glass slide by mixing 1-2 drops of freshly prepared 2% solution of sodium metabisulphite with a drop of anticoagulated blood (EDTA) and sealing the coverslip with wax or nail varnish. The preparation was kept in a covered moist petridish and observed at one hour and after 24 hours under 40X for sickle cells.
Osmotic fragility test (OFT): A 20 µl of heparinized blood was added to test tubes containing 5 ml of different concentration of sodium chloride solutions, centrifuged after mixing and leaving at room temperature for 30 minutes. Optical Density (OD) readings of the supernatant using 0.85% tube as a blank at 540 nm is taken and percentage of lysis was calculated after plotting the readings on the graph. Incubated OFT was done after incubating 2 ml of heparinized blood at 37°C for 24 hours.
Methaemoglobin reduction test was done by adding 0.5 ml of anticoagulated blood, 0.025 ml dextrose sodium nitrate solution and 0.025 ml methylene blue to a test tube, incubated for three hours at 37°C. Positive (blood and dextrose solution) and negative controls were set on, and G6PD assay was done to diagnose G6PD deficiency using standard.
Alkaline haemoglobin electrophoresis was done at pH of 8.6, by applying 3 µl of haemolysates prepared by whole blood on cellulose acetate strips, electrophoresed at 150 V for 30 minutes and were stained with Ponceau’s satin. The patterns were scanned on a scanning densitometer, and the relative percent of each band was determined.
The data was compiled on excel sheet and analysed. Since this was an observational study, only percentage calculation was applied.
Results
Over a period of five and half years, 322 cases of hereditary haemolytic anaemias were diagnosed. Spilt up of cases are summarised in [Table/Fig-1]. Haemoglobinopathies constituted majority of cases followed by hereditary spherocytosis and G6PD deficiency. A rare case like hereditary elliptocytosis and HbD Punjab were also encountered. The clinical and haematological parameters are summarised in [Table/Fig-2,3]. [Table/Fig-4] shows bands of common haemoglobins on alkaline gel electrophoresis.
Distribution of various hereditary haemolytic anaemia.
| Diagnosis | No of cases | Percentage (%) |
|---|
| Thalassaemia major | 54 | 16.45 |
| Thalassaemia trait | 91 | 28.26 |
| Thalassaemia intermedia | 20 | 6.2 |
| Sickle cell anaemia | 18 | 5.6 |
| Sickle cell trait | 5 | 1.55 |
| Sickle cell disease | 26 | 8 |
| Sickle thalassaemia | 29 | 9 |
| Hereditary spherocytosis | 43 | 13.3 |
| G6PD deficiency | 20 | 6.29 |
| Hb EE disease | 6 | 1.85 |
| HbE thalassaemia | 3 | 0.92 |
| HbE trait | 3 | 0.61 |
| Hereditary elliptocytosis | 1 | - |
| HbD punjab | 1 | - |
| Inconclusive cases | 2 | - |
Clinical parameters of various haemoglobinopathies.
| Clinical features | Thalassaemia trait | Thalassaemia intermedia | Thalassaemia major | Sickle cell trait | Sickle cell anaemia | Sickle cell- thalassaemia | Hb EE | Hb E trait | Hb E thalassaemia |
|---|
| Mean age at presentation | - | 9.16 years | 4.8 months | - | 5.5 years | 12 years | 20 years | - | 4 years |
| H/o jaundice | Nil | Nil | 9.6% | Nil | 43.7% | 66.6% | - | - | - |
| H/o transfusion | Nil | 65% | 100% | Nil | 38% | - | 20% | 50% | 100% |
| Transfusion dependency | Nil | Nil | 100% | - | - | - | - | - | 100% |
| Hepatosplenomegaly | Nil | 85% | 100% | 0 | 38% | 92% | 60% | 50% | 100% |
| Autosplenectomy | - | - | - | - | 38% | 0 | - | - | - |
Haematological parameters of haemoglobinopathies (MHBP-Microcytic Hypochromic Blood Picture).
| Haematological parameters | Thalassaemia trait | Thalassaemia intermedia | Thalassaemia major | Sickle cell trait | Sickle cell anaemia | Sickle cell-thalassaemia | Hb EE | Hb E trait | Hb E thalas- saemia |
|---|
| Hb (g/dl) | 11.8 | 6.2 | 6 | 10.8 | 7.5 | 7.7 | 9.8 | 9.5 | 5.5 |
| RBC count (million/mm3) | 5.74 | 4.2 | 2.4 | - | - | - | - | - | - |
| MCV(fl) | 62.3 | 65.3 | 69.5 | - | - | - | - | - | - |
| RDW CV% | 18.78 | 31.2 | 27 | - | - | - | - | - | - |
| Reticulocyte count% | - | 3.6 | 4.5 | 1.95 | 14.6 | 7.2 | 3 | 4 | 8.2 |
| Peripheral Boold Smear (PBS) finding | MHBP, target cells | Similar to thalassaemia major | MHBP, target cells, many nRBC’s | MHBP | Sickle cells, sickling test positive | Sickle cells, target cells sickling test positive | Many target cells | MHBP | Similar to thalassaemia major |
| Hb E/A2 % | 6.2 | 4.55 | 3.4 | - | 2.1 | 2.35 | >10 | >10 | >10 |
| Hb F% | Nil | 58.92 | 96 | - | 20 | 34 | - | | |
| Hb S% | Nil | Nil | Nil | 40 | 72.2 | 63.37 | Nil | | |
Hb electrophoresis to show locations of various haemoglobin.
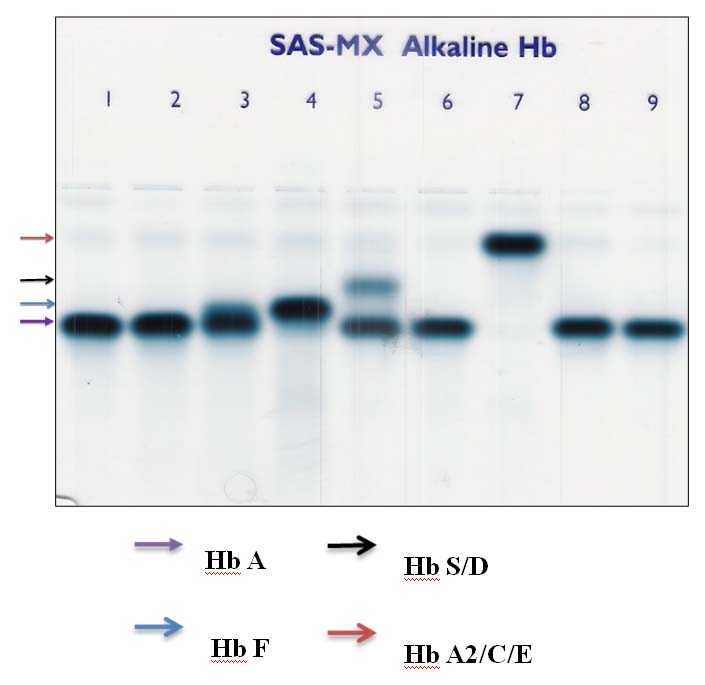
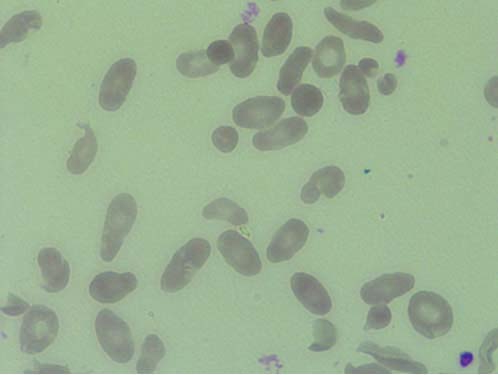
Thalassaemia trait accounted for majority of cases 91 (28.26%). The provisional diagnosis of thalassaemia trait was made based on haematological parameters like near normal Hb concentration, low red cell indices, presence of target cells in the Peripheral Blood Smear (PBS) [Table/Fig-5]. The diagnosis was confirmed by detecting elevated HbA2 levels (3.5-9%) by haemoglobin electrophoresis. In 69% of cases the diagnosis of thalassaemia trait was made incidentally when complete blood counts were requested for some other medical problems, based on PBS findings and red cell indices. In all the above cases Hb electrophoresis was suggested, but only 50% of cases were confirmed with the same. Rest of the cases of thalassaemia traits were either siblings or parents of thalassaemia major/intermedia patients. An 80% cases were less than 40 years of age at the time of diagnosis with male to female ratio of 1.75:1. Male to female ratio in thalassaemia major was 1.78:1 [Table/Fig-6]. A total of 27 patients (51.92%) were born to parents who had 2nd/3rd degree consanguineous marriage. History of transfusion prior to electrophoresis was noted in many patients, in whom HbF levels ranged from 0 to 72%. The repeat Hb electrophoresis of these patients was not possible as the transfusion free interval was less than two months. In four cases only one parent was found to be thalassaemia trait. Diagnosis of thalassaemia intermedia was made in patients who presented at later age, required blood transfusion but were not transfusion dependent and HbF levels were less than that seen in thalassaemia major.
PBS in thalassaemia trait showing crowding of RBCs with microcytic hypochromic cell and target cells (Leishman stain, 100X).
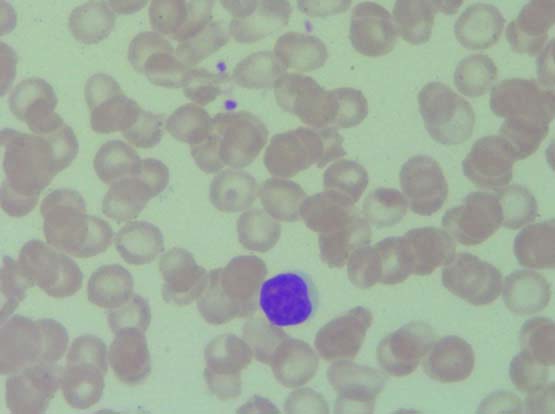
PBS in thalassaemia major showing microcytic hypochromic blood picture with cabot ring (indicated with arrow) and Nucleated RBCs (Leishman stain, 100X). (Images left to right)
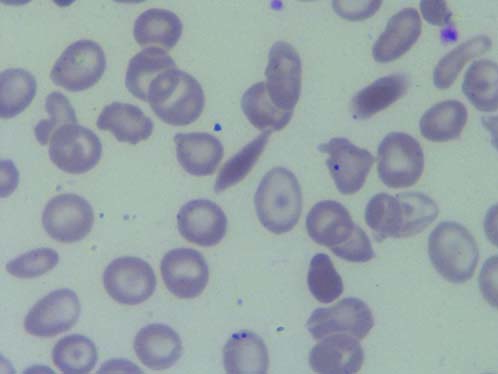
In 26 of 78 cases of sickle cell disease were not further sub grouped as Hb electrophoresis was not done. Out of 18 cases, eight were females and 10 were males. The most common clinical presentation was painful vaso-occlusive crisis. In six patients splenomegaly persisted till the age of 15 years. The diagnosis of sickle cell thalassaemia was made based on the presence of splenomegaly after childhood, family history of sickle cell trait and thalassaemia trait in the parents, PBS findings like presence of sickle cells, target cells and few nRBCs and finally on the Hb electrophoresis which showed elevated levels of HbS, HbA2, and elevated HbF [Table/Fig-7,8].
PBS showing sickle cells (Leishman stain, 100X).
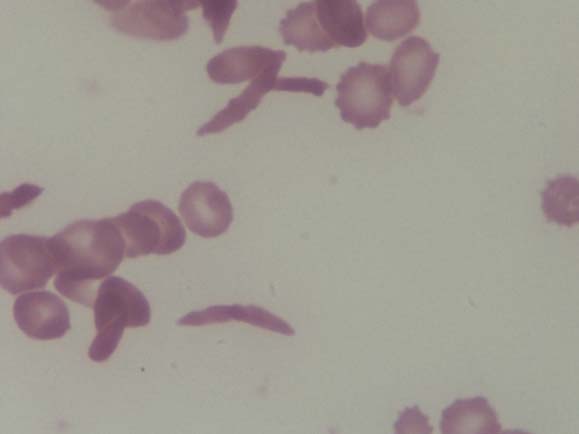
PBS showing sickle cell and target cells suggestive of sickle cell thalassaemia (Leishman stain, 100X).
A total of 11 out of 12 cases of HbE disorder were from North East India. One person, 45-year-old male was from Southern India who was adopted as a child. Hb electrophoresis: alkaline electrophoresis at pH 8.4 showed band at A2/C/E region. As the band was >10% A2 was ruled out. Acid electrophoresis was suggested to classify C/E. However, as most patients were from north east, they were provisionally diagnosed as Hb E disease.
There were 43 cases of hereditary spherocytosis with male: female ratio of 1.1:1. The age at presentation ranged widely from birth to 34 years. The presenting complaint was jaundice and cholelithiasis. On examination hepatosplenomegaly was found. Around seven patients had undergone splenectomy. Ten patients required regular transfusion. Positive family history was noted in seven cases. Average haemoglobin was 9 g/dl. Peripheral smear showed many spherocytes, microspherocytes, polychromatophils and occasional nRBCs. In all the cases RBCs showed increased osmotic fragility before and after incubation. Direct coombs test was negative in all above cases.
There were 20 cases of G6PD deficiency, 19 were males and one female. The diagnosis of G6PD deficiency was made based on the peripheral smear findings of spherocytes, bite cells, blister cells, polychromatophils and nRBCs, methaemoglobin dye reduction test and G6PD assay estimation. Most of the patients presented with jaundice (95%) and high coloured urine (60%).
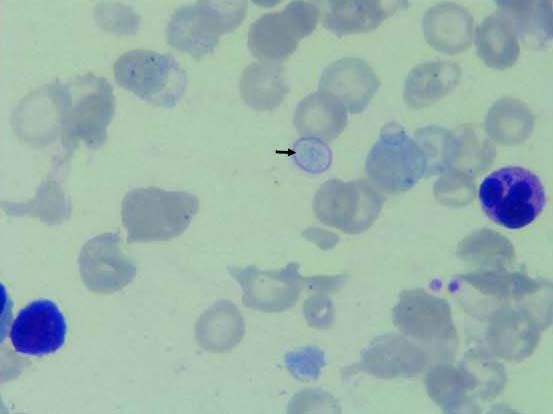
Among four miscellaneous cases one was a case of hereditary elliptocytosis. The patient presented from the age of 2.5 months, the child was born to a couple with second degree consanguineous marriage. PBS of both the parents showed many elliptocytes [Table/Fig-9]. The patient had repeated attacks of jaundice, cholelithiasis and parvovirus infection. On examination hepatosplenomegaly was found.
PBS in hereditary elliptocytosis showing many elliptocytes (>20%), (Leishman stain 100X). (Images left to right)
One case of HbD Punjab was diagnosed in a patient who came for pre-employment screening. This patient was from Punjab and alkaline haemoglobin electrophoresis showed a band at the region of HbS/D and was advised for acid electrophoresis. As sickling test was negative, a diagnosis of HbD Punjab was considered.
Discussion
Hereditary haemolytic anaemias comprise a vast and variegated spectrum of disease, some comparatively common and some very rare. Among the hereditary haemolytic anaemia haemoglobinopathies are more prevalent worldwide though some geographic areas have higher frequencies. Over 5% of the world’s population are healthy carriers of haemoglobin disorder [5]. When both the partners are carriers the risk of having a child with serious inherited anaemia is one in four with every pregnancy. Though originally endemic to the tropics and subtropics, these anaemias are now found worldwide as a result of migration. The general incidence of thalassaemia trait and sickle cell disease in India varies between 3-17% and 1-44% respectively [1,6,7]. The carrier rate of β-thalassaemia gene in Southern India varies from 1-3% and in Northern India 3-15% [8-10].
Thalassaemia: The thalassaemia syndromes were most commonly prevalent as in study done by Ambekar SS et al., [7] in Pune in contrast to study done by Balgir RS [1] in Orissa, where sickle cell disease was more prevalent. Thalassaemia trait was the most prevalent disorder among haemoglobinopathies and hereditary haemolytic anaemias. In all the incidentally detected traits on the peripheral smears, Hb electrophoresis was advised but only in 50% of cases it was done, indicating lack of awareness among the clinicians. The majority of these patients were in the reproductive age group at the time of detection of carrier state, so it is important that they know of their carrier state and necessary precautions and genetic counselling can be offered. The clinical presentation of thalassaemia major included progressive pallor in majority of cases and presented within the age of five months. Severe anaemia (<6 g/dl) and hepatosplenomegaly were found in all the cases. All the patients were transfusion dependent. History of consanguineous marriage in parents was noted in 51.92%. In these patients, awareness of the disease, premarital screening and counselling of the parents could have prevented the homozygous state. HbF levels were low in post transfusion blood samples, because of presence of normal transfused red cell. This may pose problem in definitive diagnosis if one is not aware of the transfusion history. So it is important to carry out the Hb electrophoresis to assess different variants of Hb before transfusion. The clinician needs to be informed about the need to collect sample for Hb electrophoresis before transfusion.
Thalassaemia intermedia patients have milder clinical course. There are very limited studies on thalassaemia intermedia in India and their exact prevalence is not known. The thalassaemia intermedia patients may have either heterozygous or homozygous pattern of inheritance [11]. Homozygous patients present at an early age with high HbF levels, whereas heterozygous patients present at a later age with elevated HbA2 levels and near normal HbF levels. The Hb levels of these patients ranged from 6 to 11 g/dl, few patients received transfusion infrequently and were not transfusion dependent. Their HbF levels were less than that seen in thalassaemia major patients (58.92%). All these findings are in concordance with the earlier study done by Tyagi S et al., [11]. With all the above clinical features, HbF levels and family studies, these patients can be classified as homozygous thalassaemia intermedia patients. One of the cases presented clinically like thalassaemia intermedia, requiring transfusions, had hepatosplenomegaly but his Hb electrophoresis showed mildly elevated HbA2 levels with normal HbF levels, like thalassaemia trait. This patient can be classified as heterozygous thalassaemia intermedia.
Sickle cell disease: Average frequency of sickle cell gene in India is 5%. The highest frequency of sickle cell disease in India is seen in Orissa(9%), followed by Assam (8.3%), Madhya Pradesh (7.4%), Uttar Pradesh (7.1%), Tamil Nadu (7.1%) and Gujarat (6.4%) [12]. Sickle cell trait cases in the present study were detected while screening the siblings/parents of sickle cell anaemia patients. The clinical presentation of sickle cell anaemia was milder with less severe anaemia, presence of splenomegaly up to the age of 15 years and requirement of blood transfusion only in 38% of patients, this was similar to that noted in study done by Patel AB et al., [13]. The observed high HbF levels were similar to that noted in Ponnazhagan et al., study [14]. This can explain the milder clinical course and haematological findings of the disease in this study as compared to the other studies [15,16]. Sickle cell thalassaemia disease results from inheritance of two genes, one sickle cell gene and the other thalassaemia gene. The disease may not present until late childhood, or even later, as in this study.
Hereditary Spherocytosis: Hereditary Spherocytosis is relatively common congenital haemolytic anaemia. One study done in Japan has reported hereditary spherocytosis as the most common haemolytic anaemia [17]. In this study also hereditary spherocytosis constituted second major cause of hereditary haemolytic anaemia next to haemoglobinopathies. There are few reports of hereditary spherocytosis in Indian literature [18,19]. Spherocytes can be seen in many conditions, mainly in autoimmune haemolytic anaemia and should be excluded by doing direct coombs test, which was carried out in all cases with spherocytes. The age at presentation ranged from one year to 34 years indicating variable penetrance of this disease.
G6PD deficiency: G6PD deficiency is the commonest erythrocyte enzyme deficiency disorder. Major clinical manifestation includes drug induced haemolytic anaemia [20], even here 20 of 22 patients presented with acute haemolysis after consumption of sulpha drugs for various reasons. Though drug induced haemolysis can be life threatening condition, none of the patients succumbed to death. As these patients are asymptomatic otherwise, educating them to avoid drugs and foods causing haemolysis are important in preventing haemolytic episodes.
Hb E disorder: Hb E disorder is mostly restricted to North-Eastern states of India [21]. Most of the patients in this study were from northeast India. It may be found in heterozygous form (AE), homozygous form (EE) and compound heterozygous form (e.g., Hb E with other abnormal haemoglobins or thalassaemia). The Hb EE disease was clinically milder compared to Hb E thalassaemia disorder, with later age of presentation (20 years), milder clinical course like chronic anaemia, recurrent abdominal pain, generalised weakness and low transfusion requirement. The HbE thalassaemia disorder resembled thalassaemia major/intermedia, with early age of presentation, progressive pallor, hepatosplenomegaly, PBS showing microcytic hypochromic blood picture with target cells and presence of many nRBCs. All the above features were consistent with the earlier studies [21,22].
One case of hereditary elliptocytosis was diagnosed. The definitive diagnosis mainly depends on the morphology on the PBS, their percentage and the screening of the parents. HbD Punjab is commonly encountered in Punjab state [23], the present one case of HbD Punjab was also from the same region.
Presently newer, more sensitive methods to detect abnormal haemoglobins are available like Cation-Exchange High-Performance Liquid Chromatography (CE-HPLC), capillary electrophoresis and are good alternative to haemoglobin electrophoresis. Electrophoretic studies like cellulose-acetate electrophoresis or isoelectric focusing are still in practice in some of the centres. Naked Eye Single Tube Red Cell Osmotic Fragility Test (NESTROFT) is used as first screening test in many developing countries including India, as it is cost effective, rapid and reliable [24]. All these methods have their own advantages and disadvantages in detecting β-thalassaemia trait, common (HbS) and rare haemoglobin variants (HbC, unstable α chain variants). In resource poor centres complete blood count, PBS examination and simple tests to detect abnormal haemoglobins still holds good for screening.
Management of thalassaemia major, a common haemoglobin disorder is by regular blood transfusion. So the affected child is at risk of transfusion transmitted diseases and side effects of iron overload. The only curative therapy available is stem cell transplantation which costs around 8-10 lacs, which many of the families in developing countries cannot afford [9]. Future perspective is to aim at prevention of occurrence of thalassaemia major. This involves screening for carriers, genetic counselling (premarital and preconception) and prenatal diagnosis in high risk couples by PCR based methods like reverse dot-blot hybridisation, amplification refractory mutation system and DNA sequencing on chorionic villous sampling. Few centres perform HPLC on cord blood in second trimester [25].
The present study though old is still relevant as the prevalence of these disease has not changed much. It is comprehensive, descriptive study explaining the different hereditary haemolytic anaemias prevalent in the region and helps to give a picture of them in this part of India.
Limitation
As it is a hospital based study it has few limitation. It may not give accurate prevalence of each disease. Being referral hospital rare cases are referred from other regions like HbE is also represented.
Conclusion
Hereditary haemolytic anaemias have a particular pattern of inheritance and thus transmitted to the offspring. Haemoglobinopathies which constituted the major group of hereditary haemolytic anaemia has a great burden on the families and the society. Stem cell transplantation is the only available curative treatment which cannot be afforded by many families. Hence prevention plays an important role in bringing down the incidence of these disorders. Screening for thalassaemia minor and genetic counselling of people found positive is one of the important steps in achieving this. According to the present study, awareness among clinicians about thalassaemia trait is low, educating all the medical fraternity might help reduce the incidence of thalassaemia major. More than half of thalassaemia major patients were offsprings of consanguineous marriage, this highlights the need to diagnose thalassaemia trait and provide premarital/prenatal counselling.