Limb Girdle Muscular Dystrophy (LGMD): Case Report
Shubhangi A. Kanitkar1, Meenakshi Kalyan2, Anu N. Gaikwad3, Ankit Makadia4, Harshad Shah5
1 Professor, Department of Medicine, Padmashree Dr. D. Y. Patil Medical College Hospital and Research Centre, Pimpri, Pune, India.
2 Associate Professor, Department of Medicine, Padmashree Dr. D. Y. Patil College Hospital and Research Centre, Pimpri, Pune, India.
3 Professor, Department of Medicine, Padmashree Dr. D. Y. Patil College Hospital and Research Centre, Pimpri, Pune, India.
4 Chief Resident, Department of Medicine, Padmashree Dr. D. Y. Patil College Hospital and Research Centre, Pimpri, Pune, India.
5 Senior Resident, Department of Medicine, Padmashree Dr. D. Y. Patil College Hospital and Research Centre, Pimpri, Pune, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Meenakshi Kalyan, RH no. 24, Mayureshwar, Sai Nisarg Park, Pimple Saudagar, Pune-411027, India. E-mail : drmeenakshi.kalyan@gmail.com
We report a young male of autosomal recessive limb girdle muscular dystrophy (LGMD) with positive family history presented with gradual onset proximal muscle weakness in all four limbs since eight years and thinning of shoulders, arms and thighs. Neurological examination revealed atrophy of both shoulders with wasting of both deltoids thinning of thighs and pseudo hypertrophy of both calves, hypotonia in all four limbs. Gower’s sign was positive. Winging of scapula was present. Power was 3/5 at both shoulders, 4/5 at both elbows, 5/5 at both wrists, 3/5 at both hip joints, 3/5 at both knees, 5/5 at both ankles. All deep tendon reflexes and superficial reflexes were present with plantars bilateral flexors. Electromyography (EMG) showed myopathic pattern. He had elevated creatinine phosphokinase levels and muscle biopsy findings consistent with muscular dystrophy.
Gower’s sign, Muscle biopsy, Proximal muscle weakness
Case Report
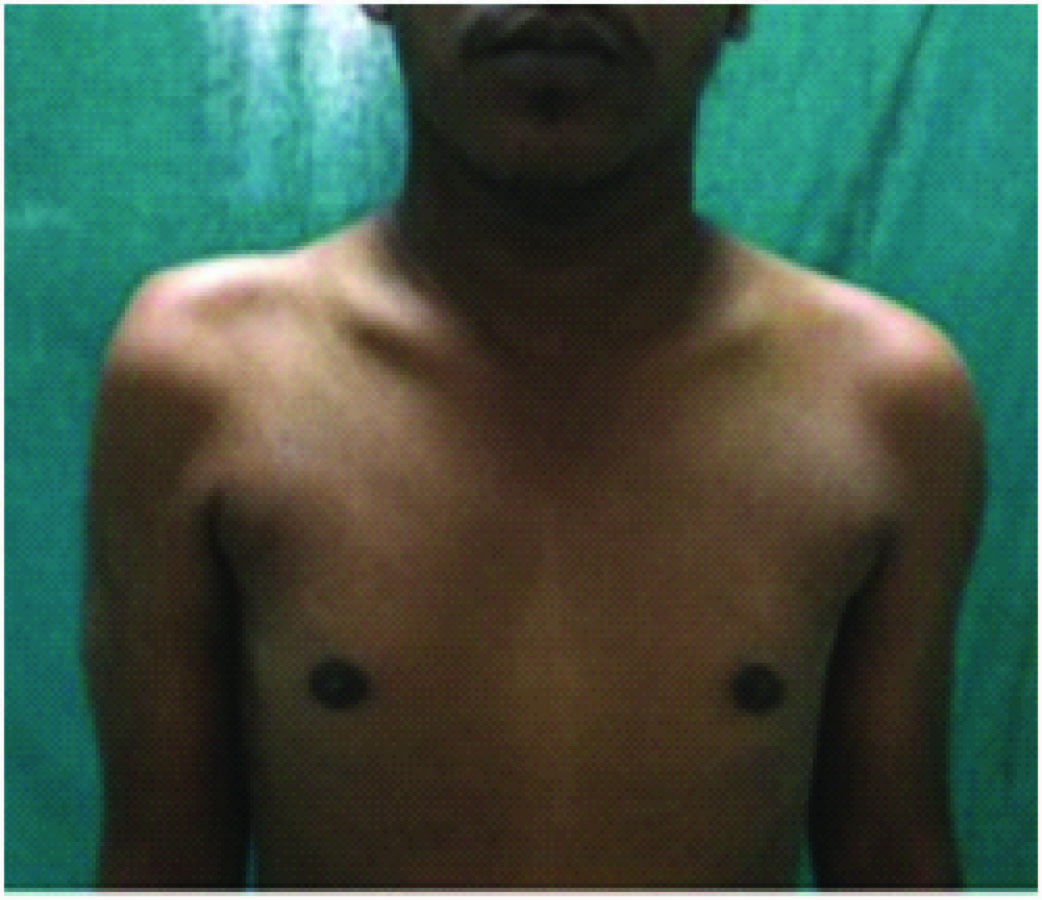
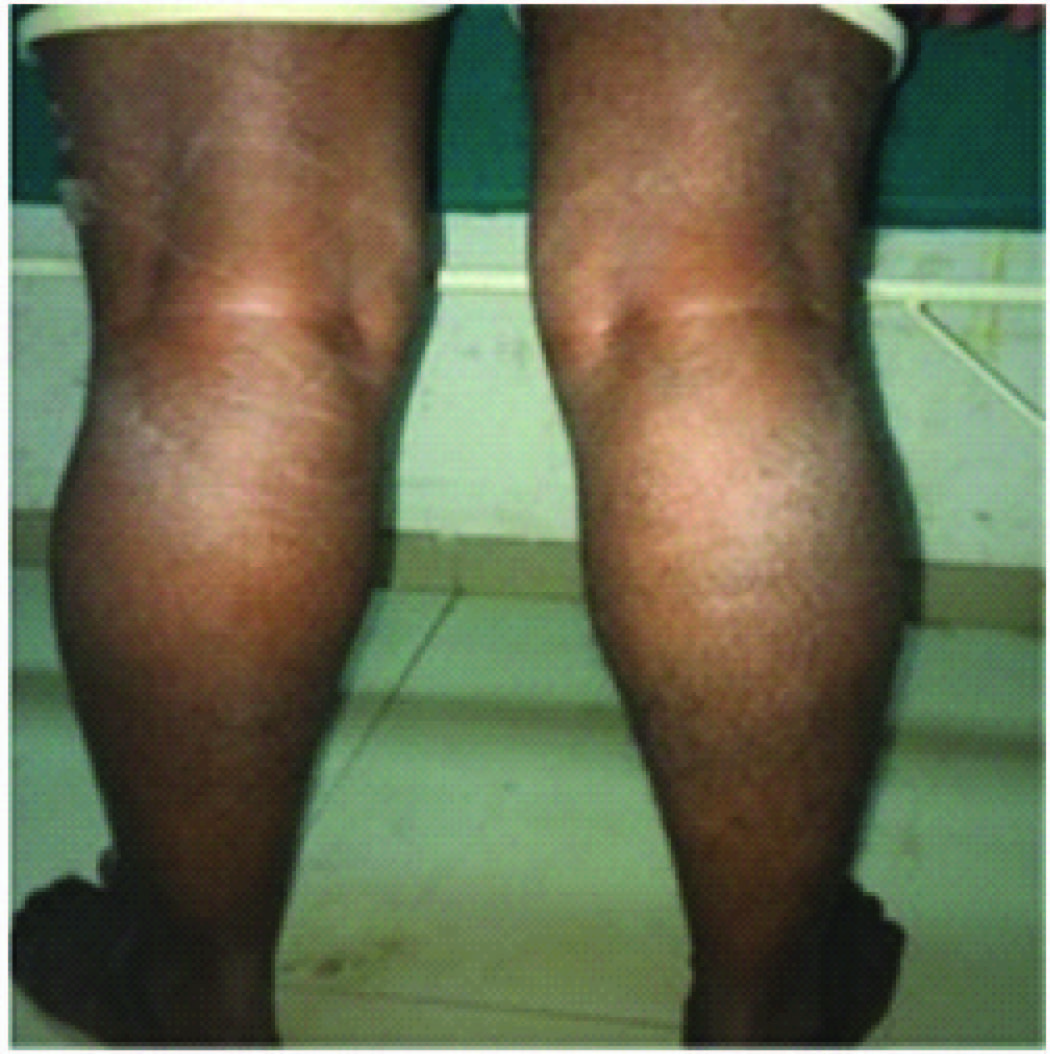
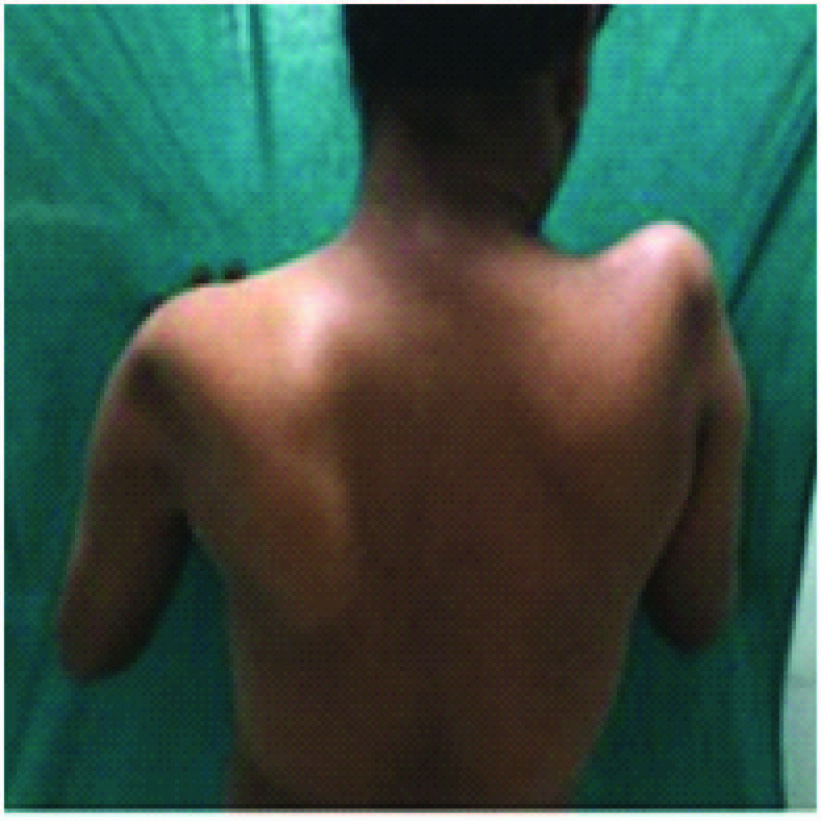
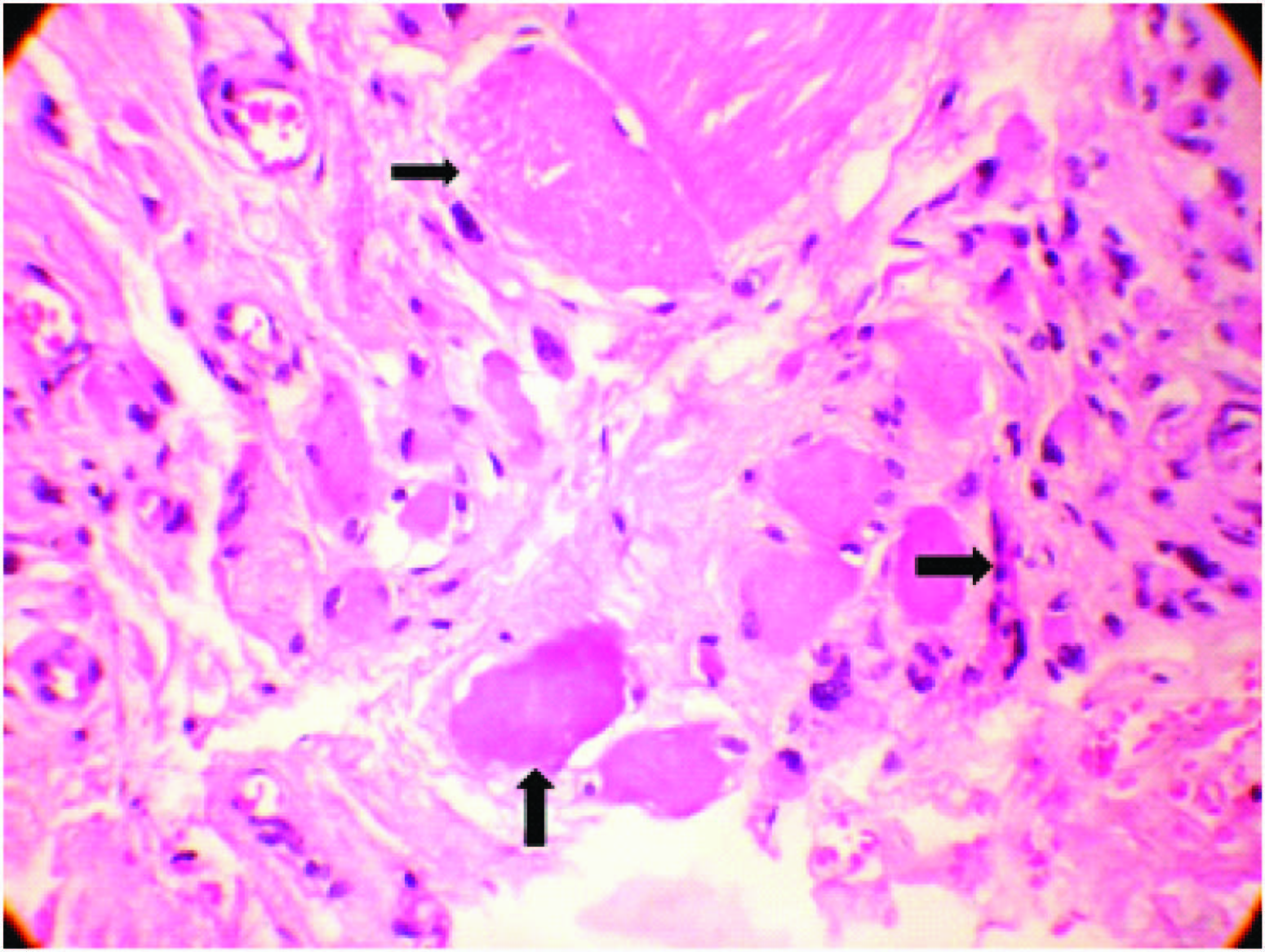
A 26-year-old male presented with proximal weakness in both upper limbs and lower limbs since eight years, which was insidious in onset and gradually progressive. He had difficulty in climbing stairs, running, getting up from squatting position, and raising arms above the head. There was thinning of shoulders, arms and thighs. No history of distal muscle weakness, twitching of the muscles, pain in the limbs, breathlessness, cranial nerve involvement or sensory symptoms and no bladder, bowel disturbances. No history of addictions or drug intake. He is the third child born of non-consanguineous marriage. Birth history and developmental milestones were normal. Family history revealed that his elder sister who is 30-year-old, suffered from similar weakness in limbs since 12 year and is now wheel chair bound. General physical examination was normal with single breath count of 28. Neurological examination showed higher mental functions including speech and cranial nerves were normal. He had atrophy of both shoulders with wasting of both deltoids [Table/Fig-1], thinning of thighs and pseudo hypertrophy of both calves [Table/Fig-2], hypotonic in all four limbs. Power was 3/5 at both shoulders, 4/5 at both elbows, 5/5 at both wrists, 3/5 at both hip joints, 3/5 at both knees, 5/5 at both ankles. Gower’s sign was positive. Winging of the scapula was present [Table/Fig-3]. All deep tendon reflexes and superficial reflexes were present with plantars bilateral flexors. Sensory system was normal. He had waddling gait. There were no cerebellar signs, skull and spine was normal. Respiratory, cardiovascular and abdominal system was unremarkable. Investigations showed haemogram, liver function, renal function tests and serum electrolytes, thyroid function tests, s. cortisol, s. LDH, s. PTH were normal. Creatinine phosphokinase was 1570 IU/l. (Reference: 24-190 IU/l). Urine routine and urine for myoglobin was negative. ECG, chest X-ray, echocardiography, and USG abdomen was normal. Nerve conduction studies were normal. EMG performed in right upper and lower limb showed myopathic pattern. Muscle biopsy from right quadriceps muscle showed muscle fibres of various sizes, fibre necrosis and fibre regeneration. Large hyaline hypereosinophilic fibres scattered within the vesicles. There was crowding of the nuclei in the centre, along with multinucleation, degeneration of fibres and fat infiltration consistent with muscular dystrophy [Table/Fig-4]. Genetic studies and DNA analysis was not possible due to financial constraints. Based on the history, proximal muscle weakness, creatinine phosphokinase levels, EMG and muscle biopsy, diagnosis of LGMD was made.
Atrophy of both shoulders with wasting of both deltoids
Pseudo hypertrophy of calves
Photomicrograph of muscle biopsy with haematoxylin and eosin staining showing fibre necrosis and regeneration with large hyaline hypereosinophilic fibres scattered within the vesicles, crowding of the nuclei in the centre, multinucleation, degeneration of fibres and fat infiltration with magnification 40 X
Discussion
LGMDs are a heterogeneous group of genetically determined by progressive disorders of skeletal muscle with a primary or predominant involvement of the pelvic and shoulder girdle musculature. Because of their heterogeneity and the lack of diagnostic specificity, there are few reports on the prevalence of LGMD. Estimates for prevalence of all forms of LGMD range from one in 14,500 to one in 123,000 [1]. Reports on LGMDs and their subtypes are very few from India [2].
The syndrome of LGMD represents more than one disorder. Both males and females are affected with onset ranging from late in the first decade to the fourth decade. Respiratory insufficiency from weakness of the diaphragm and cardiomyopathy may occur. A systemic classification is based on autosomal dominant (LGMD 1) and autosomal recessive (LGMD 2) inheritance. Presently there are seven autosomal dominant and 10 autosomal recessive disorders [Table/Fig-5] [3]. Differential diagnosis include adult variant of spinal muscular atrophy (SMA III, Kugelberg-Welander disease), polymyositis, dermatomyositis, other musculardystrophies e.g., facio-scapulo-humeral, Becker, Duchenne muscular dystrophy, endocrine and acquired metabolic myopathies (e.g., Cushing’s disease, hyperthyroidism, steroid, and statin administration). Santra [4] reported a sporadic case of LGMD in an adult female who had contractures of shoulder girdle muscles and normal creatinine phosphokinase. Our patient is autosomal recessive type of LGMD who had pseudo hypertrophy of the calves and raised creatinine phosphokinase.
Autosomal recessive LGMDs
| Disease | Clinical features | Locus or gene |
|---|
| LGMD 2A | Onset 1st or 2nd decade, Tight heel cords, contractures at elbows, wrists, fingers, rigid spine, proximal and distal weakness | Calpain 3 |
| LGMD 2B | Onset 2nd or 3rd decade, proximal muscle weakness at onset, later calf muscles affected. Miyoshi myopathy is variant of LGMD 2B with calf muscles affected at onset [3] | Dysferlin |
| LGMD 2C-F | Onset in childhood to teenage. Clinical condition similar to Duchenne and Becker muscular dystrophies, cardiomyopathy uncommon, cognitive function normal. | sacroglycans |
| LGMD 2G | Onset age 10-15 yrs, proximal and distal muscle weakness [3] | Telethonin |
| LGMD 2H | Onset 1st to 3rd decade, proximal muscle weakness | TRIM 32 gene |
| LGMD 2I | Onset 1st to 3rd decade. Clinical condition similar to Duchenne or Becker dystrophies, cardiomyopathy, cognitive function normal. | Fukutin related protein |
| LGMD 2J | Onset 1st to 3rd decade, proximal lower limb weakness, mild distal weakness, progressive weakness causes loss of ambulation | Titin |
| LMGD 2K | Usually presents in infancy as Walker Wanrburg syndrome but can present in early adult life with proximal weakness and only minor CNS abnormalities | POMT 1 |
| LGMD 2IL | Presents in childhood or adulthood. May manifest with quadriceps atrophy and myalgia. Some present with early involvement of the calves in the second decade of life, resembling Miyoshi myopathy | Anoctamin 5 |
| LGMD 2M | Usually presents in infancy as Fukuyuma congenital muscular dystrophy but can present in early adult life with proximal weakness and only minor CNS abnormalities | Fukutin |
| LGMD 2 N | Usually presents in infancy as Muscle-Eye-Brain disease but can present in early adult life with proximal weakness and only minor CNS abnormalities | POMGn T1 |
| LGMD 2O | Usually presents in infancy as Walker Warburg syndrome but can present in early adult life with proximal weakness and only minor CNS abnormaliies | POMT2 |
The gold standard for secure diagnosis is the demonstration of causative mutations in the relevant gene. Because of the large number of loci involved, however, it is generally necessary to direct mutation detection to specific loci. Combined analysis using both immunoblotting and immunocytochemistry using a full range of diagnostic antibodies improves the diagnostic yield. The diagnostic process therefore needs to integrate clinical analysis, protein testing and genetic analysis, which is focused in specialized centres [5]. Jethwa et al., [6] reported LGMD type 2B autosomal recessive inherited type masquerading as inflammatory myopathy. Our patient had no pain in the limbs and genetic analysis could not be performed due to financial constraints. Supportive care should be offered for neuromuscular disabilty, including ambulatory aids if necessary. Stretching of contractures is difficult. Management of cardiomyopathy and arrhythmias can save lives [3]. Prognosis depends on the type of LGMD and whether there is cardiac or respiratory involvement. In general, all types of LGMD progress with time, but this is highly variable between the different LGMD types, and also between individuals with the same specific type or within a family.
Conclusion
Our case describes autosomal recessive LGMD with positive family history, elevated creatinine phosphokinase and diagnostic muscle biopsy. Onset,progression and distribution of the weakness and wasting vary considerably among individuals and genetic subtypes.
[1]. Urtasun M, Saenz A, Roudaut C, Poza JJ, Urtizberea JA, Cobo AM, Limb-girdle muscular dystrophy in Guipuzcoa (Basque Country, Spain)Brain 1998 121:1735 [Google Scholar]
[2]. Meena AK, Sreenivas D, Sundaram C, Sarcoglycanopathies: A clinico-pathological studyNeurol India 2007 55:117-21. [Google Scholar]
[3]. Amato Anthony A, Brown Robert H., Jr, Muscular dystrophies and other muscle diseases. In Longo, Fauci, Kasper, Hauser, et al. EditorHarrison’s Principles of Internal medicine 2012 18(2):3493 [Google Scholar]
[4]. Gouranga Santra, Limb girdle muscular dystrophyJIACM 2008 9(4):302-05. [Google Scholar]
[5]. Laval SH, Bushby KMD, Limb girdle muscular dystrophy from genetics to molecular pathologyNeuropathology and Applied neurobiology 2004 30:91-105. [Google Scholar]
[6]. Jethwa H, Jacques TS, Gunny R, Wedderburn LR, Pilkington C, Manzur AY, Limb girdle muscular dystrophy type 2B masquerading as inflammatory myopathy: case reportPediatr Rheumatol online J 2013 11(1):19 [Google Scholar]